Category: Genetics (Non-PD)
Objective: To describe the clinic-genetic profile of clinically suspected mitochondrial cytopathies with movement disorders (MDs).
Background: Literature on the clinico-genetic spectrum of movement disorders related to mitochondrial cytopathies is sparse.
Method: We performed a retrospective chart review of all cases of clinically suspected mitochondrial cytopathies based on mitochondrial disease criteria (MDC 2006).1 Data were extracted with regards to the clinic-investigational profile and genetics based on whole exome sequencing (WES).
Results: We identified 76 cases (male=43, 57.3%) of clinically suspected mitochondrial cytopathies, with a median age at onset (AAO) of 10 years [IQR 3-19, range 1 month-45 years], and presentation of 15 years [(IQR 6-25, (1.5-51 years)]. The majority of cases (n=36,73.5%) belonged to the possible group based on MDC 2006. A total of 49 cases (65.3%) manifested MDs (males=27,55.1%). The median AAO of MD was 12 years [IQR 3.6 to 20 (0.25-45) years], while MD at onset was observed in 28 cases (57.1%). The spectrum of MDs comprised ataxia (n=32,65.3%), dystonia [n=18,36.7%; focal=8(44.4%),generalized=6,(33.3%),multifocal=3(16.7%),and segmental=1,5.6%)], tremors (n=10, 20.4%), recurrent falls (n=9,18.4%), parkinsonism (n=8,16.3%), myoclonus (n=8,16.3%) and chorea (n=4,8.2%). The primary MD symptom initially seen was cerebellar ataxia(n=25,51%). The common associated systemic symptoms included seizures (n=14,28.6%), neuropathy (n=11,22.4%), hearing loss (n=11,22.4%), and optic atrophy (n=9,18.4%). WES yielded genetic diagnoses in 16% overall (n=12/75), and 16.3% (n=8/49) amongst the MD cases. Predictors for genetic positivity included AAO<10 years in pediatric cases (p=0.010) and brain MRI indicating mitochondrial etiology (p<0.001).[Figure 1] The presence of MD was not significantly linked with genetic positivity (p>0.99). The genetic landscape in the MD subgroup comprised mitochondrial complex deficiencies (n=5/8,62.5%) followed by Leigh spectrum (n=1/8,12.5%) and pyruvate dehydrogenase E2 deficiency ((n=1/8,12.5%). Four cases had a negative genetic report despite a positive muscle biopsy. Non-mitochondrial genetic diagnoses were found in three cases: Krabbe’s disease, SYNE1-related ataxia, and Charcot Mary Tooth 4B.
Conclusion: MDs related to mitochondrial cytopathies represent a unique spectrum that require a high index of suspicion for prompt diagnosis and management.
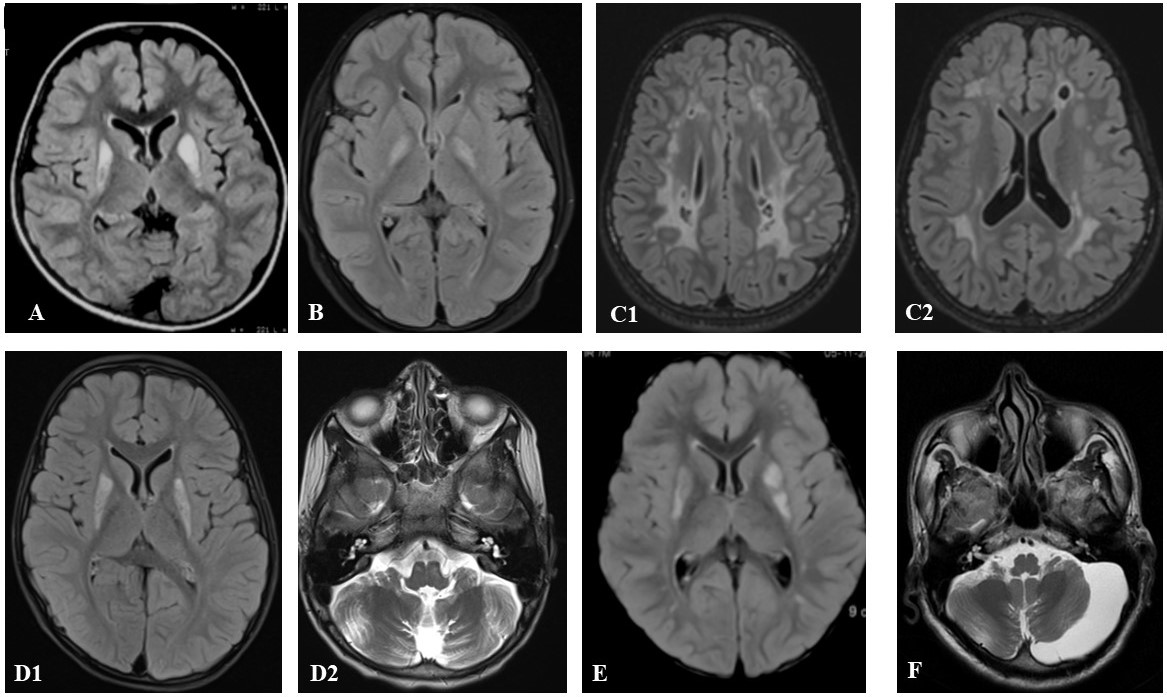
Brain MRI findings of genetically proven cases
References: Morava E, Van Den Heuvel ; L, Hol ; F, et al. Mitochondrial disease criteria Diagnostic applications in children [online]. 2006. Accessed at: www.neurology.org.
To cite this abstract in AMA style:
D. Dhar, V. Holla, P. Phulpagar, R. Yadav, N. Netravathi, B. Muthusamy, P. Pal. Utility of Whole Exome Sequencing in movement disorders potentially related to Mitochondrial cytopathies [abstract]. Mov Disord. 2024; 39 (suppl 1). https://www.mdsabstracts.org/abstract/utility-of-whole-exome-sequencing-in-movement-disorders-potentially-related-to-mitochondrial-cytopathies/. Accessed April 26, 2025.« Back to 2024 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/utility-of-whole-exome-sequencing-in-movement-disorders-potentially-related-to-mitochondrial-cytopathies/