Session Information
Date: Tuesday, June 6, 2017
Session Title: Rare Genetic and Metabolic Diseases
Session Time: 1:45pm-3:15pm
Location: Exhibit Hall C
Objective: To illustrate phenotype of PLP1-related disorder caused by a novel mutation.
Background: Phenotypes of X-linked PLP1-related disorders vary from severe forms of hypomyelinating leukodystrophy-Pelizaeus–Merzbacher disease, clinically characterized by nystagmus, spastic quadriplegia, ataxia, dystonia and developmental delay, to mild forms of hereditary spastic paraplegia type 2.
Methods: Patient with an unremarkable family history was born following uncomplicated pregnancy and delivery, but was floppy baby with delayed psychomotor milestones. He achieved independent walking at the age of 23 months, however, his gait was spastic and ataxic. Over the time gait slowly worsened: at the age of 12 he needed walking stick and was wheelchair bound at the age of 20. The first dystonic feature was writer’s cramp observed early at primary school. Thereafter, dystonia slowly generalized. Vision has been poor from childhood. He finished primary school with personal assistance help. Parents noticed significant progression of cognitive decline and behavioural disturbances. From the age of 16 he has had urinary urgency. Brain CT at the age of 3 revealed global but mild atrophy of cerebral cortex and vermis. He was diagnosed with mild optic atrophy at age of 4. On the last examination (26 years), he had vertical gaze palsy (particularly on upward gaze), pronounced delay on saccadic initiation, head and voice tremor, dysarthria, generalized dystonia, spastic paraplegia, and stimulus sensitive myoclonus. Intention tremor was present on finger-to-nose test. Somatic examination was unremarkable. Interestingly enough, nystagmus has never been observed.
Results: Brain MRI revealed diffuse hypomyelination, global brain atrophy, thin corpus callosum and atrophy of the cerebellum, especially vermis [figure1]. A hemizygous variant in the PLP1 gene, c.354del (p.Gly120Alafs*27) was detected. This new mutation creates a shift in the reading frame starting at codon Gly120. The new reading frame ends in a STOP codon 26 positions downstream.
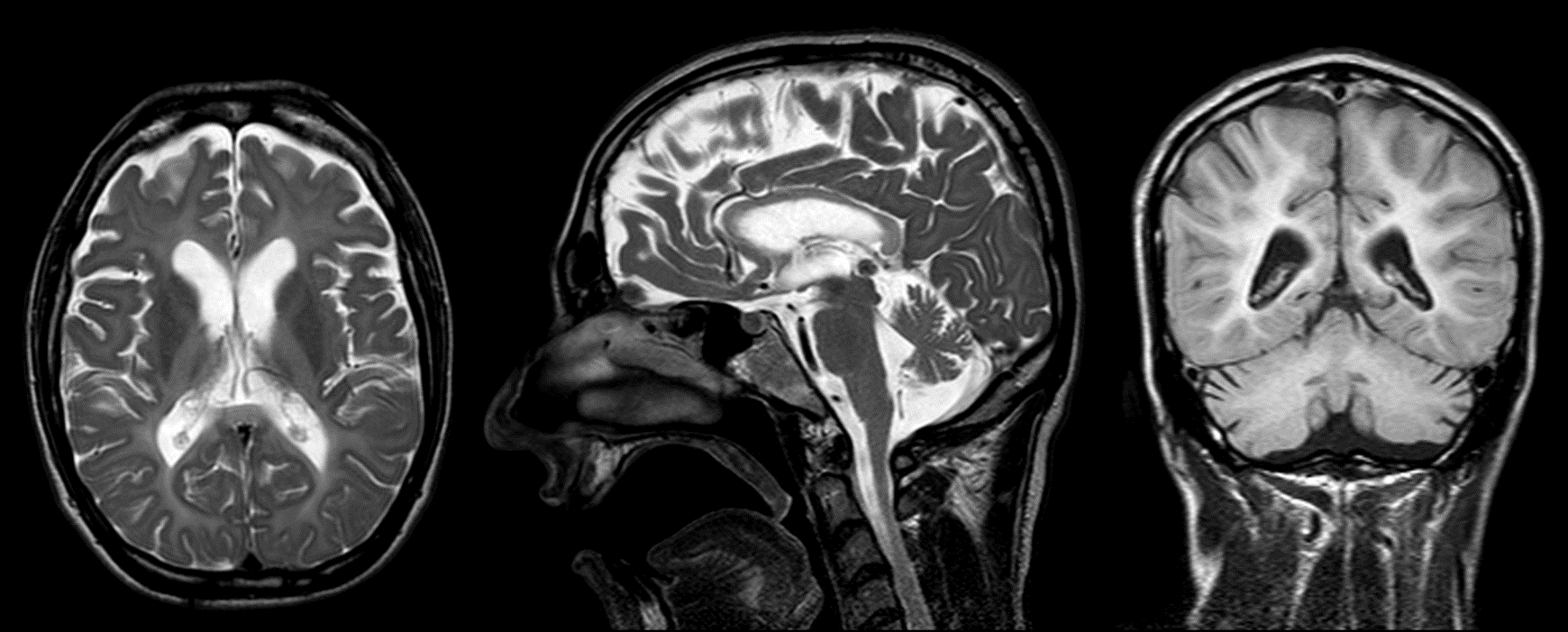
Conclusions: We reported a patient with new PLP1 mutation, characterized, in addition to typical findings, with the presence of stimulus sensitive myoclonus and vertical gaze palsy, lack of nystagmus during the disease course and cerebellar atrophy on MRI examination.
To cite this abstract in AMA style:
N. Kresojevic, I. Petrovic, V. Dobricic, A. Tomic, M. Svetel, V. Kostic. Phenotype of PLP1-related disorder caused by novel mutation: a case report [abstract]. Mov Disord. 2017; 32 (suppl 2). https://www.mdsabstracts.org/abstract/phenotype-of-plp1-related-disorder-caused-by-novel-mutation-a-case-report/. Accessed April 21, 2025.« Back to 2017 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/phenotype-of-plp1-related-disorder-caused-by-novel-mutation-a-case-report/