Category: Parkinson's Disease: Genetics
Objective: Integrate proteomics from the cerebrospinal fluid (CSF) and urine from the Parkinson’s Progression Markers Initiative cohort to prioritize the risk variants associated with Parkinson’s disease (PD).
Background: There are ~ 90 loci associated with PD risk, including common variants in loci near the GBA, SNCA, LRRK2, PGRN, CTSB, and GPNMB genes. The causal mediators of phenotype-associated genetic variation remain unclear.
Method: A total of 1003 (846) individuals passed genome-wide association QC steps, including 562 (436) PD cases, including 139 (133) LRRK2 and 61 (71) GBA carriers, and 355 (232) sporadic PD (sPD) cases, 171 (132) controls and 270 (278) prodromal cases with 5k CSF (urine) proteomics, integrating with 5M genetic variants across whole genome. We identified protein quantitative trait loci (pQTLs) using PLINK2.0[1] and causal proteins in CSF and urine, leveraging Mendelian randomization[2] and colocalization[3]. Proteomic signatures were curated by comparing proteomics across different mutation types, adjusted for age at baseline, sex, and the first four principal components. Using an elastic logistic regression model to predict PD risk. We implemented weighted gene co-expression network analysis (WGCNA)[4] to identify subclusters with distinct molecular networks. We will use proteomic and genomic data from the Parkinson’s Disease Biomarkers Program (PDBP) cohort as independent replication.
Results: We found 824 (153) cis-pQTLs and 500 (11) trans-pQTLs for CSF (urine) proteomics; 47 (4) causal proteins, and two proteins (ARSA and HP) common to both CSF and urine; 12 (zero) colocalizing with PD risk loci. We identified CSF proteins associated with LRRK2 (281 proteins), GBA (132 proteins), sPD (54 proteins). A model with LRRK2-associated proteins can distinguish PD from sPD (AUC = 0.95), and a model with sPD-associated proteins can distinguish sPD and Control (AUC = 0.837). We uncovered two sPD subclusters that exhibit different CSF levels of p-tau (p = 1.3×10-13), t-tau (p = 6.5×10-15), a-syn (p = 4.4×10-11), and a-beta (p = 1.4×10-7) [figure 1,2a-d].
Conclusion: Integrating proteomics and genomics data identifies candidate risk variants and potential causal mediator proteins in PD. Moreover, combining multiple proteomic signatures uncovers two sPD subclusters with distinctive changes in neurodegenerative-associated proteins.
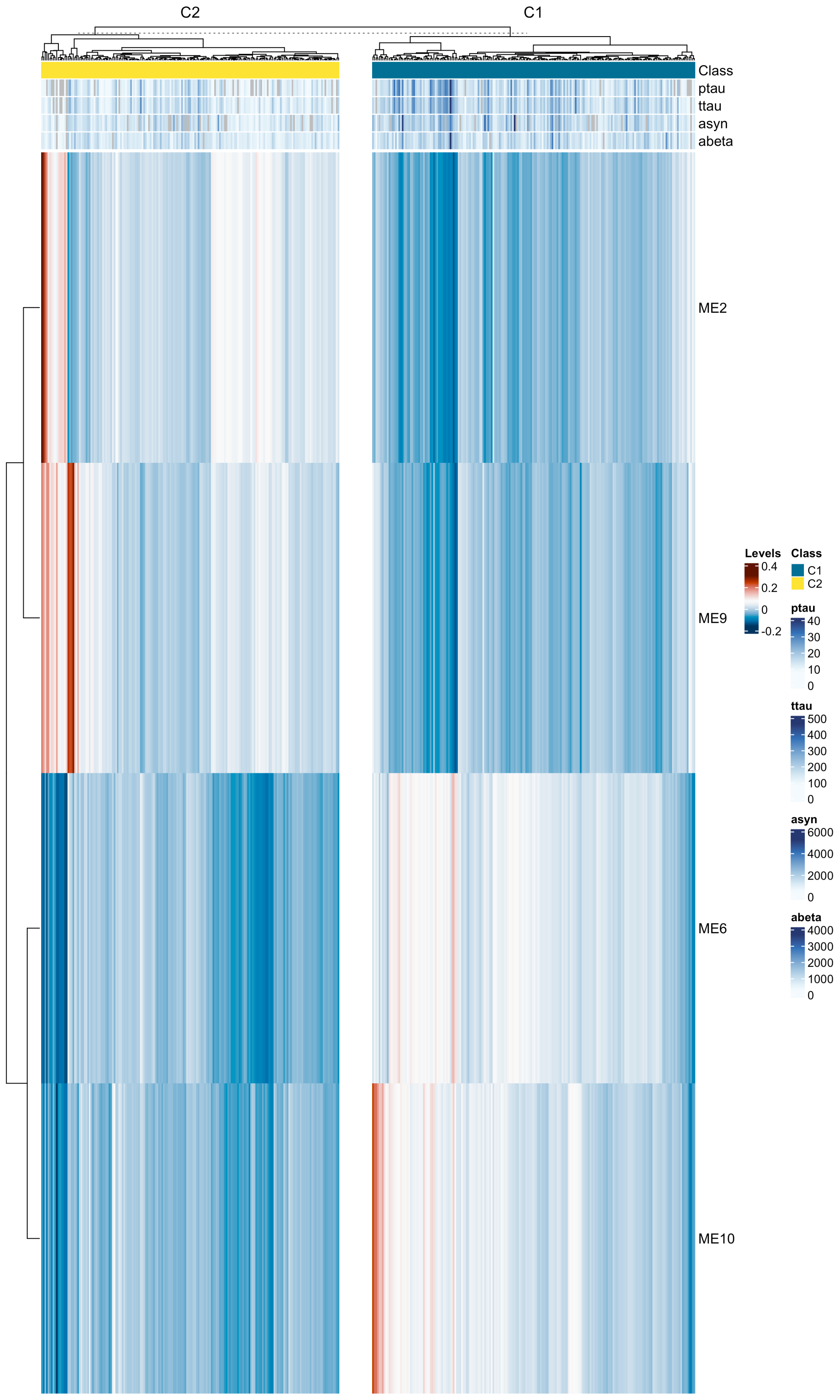
Heatmap of sPD subtypes
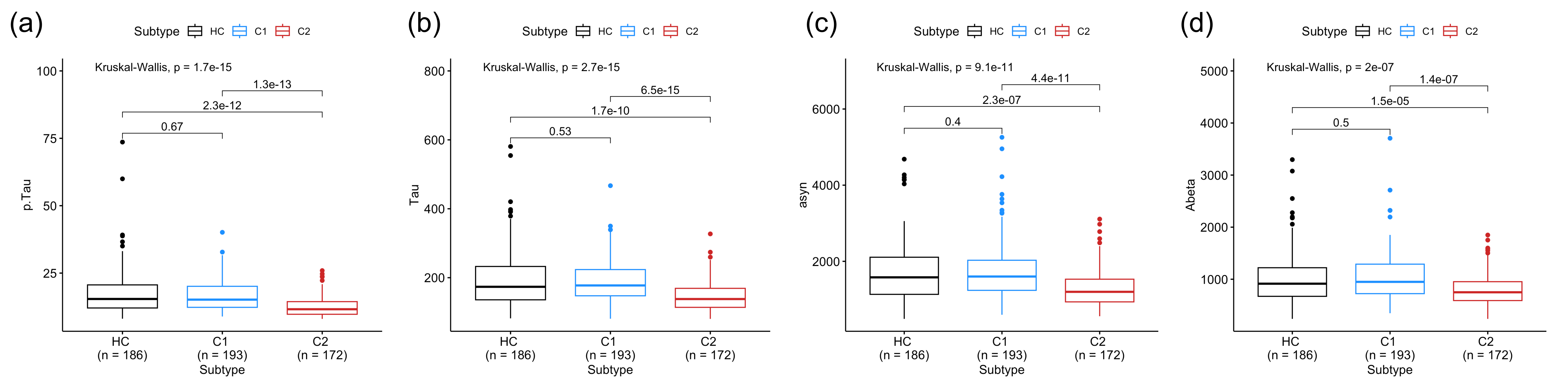
Comparisons of CSF protein levels
References: 1. Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., & Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience, 4(1), s13742-015.
2. Hemani, G., Zheng, J., Elsworth, B., Wade, K. H., Haberland, V., Baird, D., … & Haycock, P. C. (2018). The MR-Base platform supports systematic causal inference across the human phenome. elife, 7, e34408.
3. Giambartolomei, C., Vukcevic, D., Schadt, E. E., Franke, L., Hingorani, A. D., Wallace, C., & Plagnol, V. (2014). Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS genetics, 10(5), e1004383.
4. Langfelder, P., & Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics, 9, 1-13.
To cite this abstract in AMA style:
M. Lai, B. Benitez. Leveraging Large-Scale Proteomics to Identify Protein Quantitative Trait Loci and Causal Effects on Parkinson’s Disease [abstract]. Mov Disord. 2024; 39 (suppl 1). https://www.mdsabstracts.org/abstract/leveraging-large-scale-proteomics-to-identify-protein-quantitative-trait-loci-and-causal-effects-on-parkinsons-disease/. Accessed June 14, 2026.« Back to 2024 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/leveraging-large-scale-proteomics-to-identify-protein-quantitative-trait-loci-and-causal-effects-on-parkinsons-disease/