Category: Other
Objective: To report the case series of patients with late-onset Wilson’s disease (WD) with predominantly neurological manifestation.
Background: WD is a rare and treatable disorder of copper metabolism with an autosomal recessive inheritance due to mutations in the ATP7B gene. WD is characterized by excessive deposition of copper in the liver, brain and other tissues. The hepatic and neurological forms of WD have wider variations. WD usually presents in children, adolescents, and young adults. Late-onset WD forms diagnosed after age 40 years are rarely reported.
Method: Clinical presentation, laboratory tests, brain MRI study and mutation analysis were evaluated in seven patients with late-onset WD.
Results: In our cohort of 49 adult WD patients, late-onset WD was identified in 7 individuals (14.3%) [Table 1]. Median age was 43 years (range 40–59), age of onset – 41 years (range 40–45), male/female – 3/4. Five patients presented with neurological debut of the disease, in one case the first manifestation was bipolar disorder and one patient was asymptomatic (patient’s sibling). Six patients presented various neurological symptoms: parkinsonism – 6/6, tremor – 5/6, cerebellar ataxia – 5/6, pyramidal signs – 4/6, dystonia – 3/6, dysarthria – 3/6, dysphagia – 2/6, upward gaze paresis – 2/6, chorea – 1/6, myoclonus – 1/6, stereotypy – 1/6. Six patients had liver disease (сirrhosis – 3, fibrosis – 3). Laboratory tests revealed low platelet count (3/7), alanine transferase and/or aspartate transferase elevation (2/7), low of serum copper (5/7), low ceruloplasmin level (6/7), elevation of 24-hour urinary copper excretion (7/7). Kayser-Fleischer rings were found in 6/7 patients. Six patients presented brain MRI changes: brainstem – 6/6, сerebellum – 6/6, thalamus – 3/6, and basal ganglia – 2/6 patients. Six patients were homozygous for p.H1069Q mutation (85,7% of all alleles), another rare pathogenic variant was c.2121+3A>T (rs1248002612).
Conclusion: In our series of late-onset WD patients had more neurological than hepatic manifestations with a wide range of movement disorders. Most patients were homozygous for “slavic” p.H1069Q mutation which may determine the phenotype of the disease.
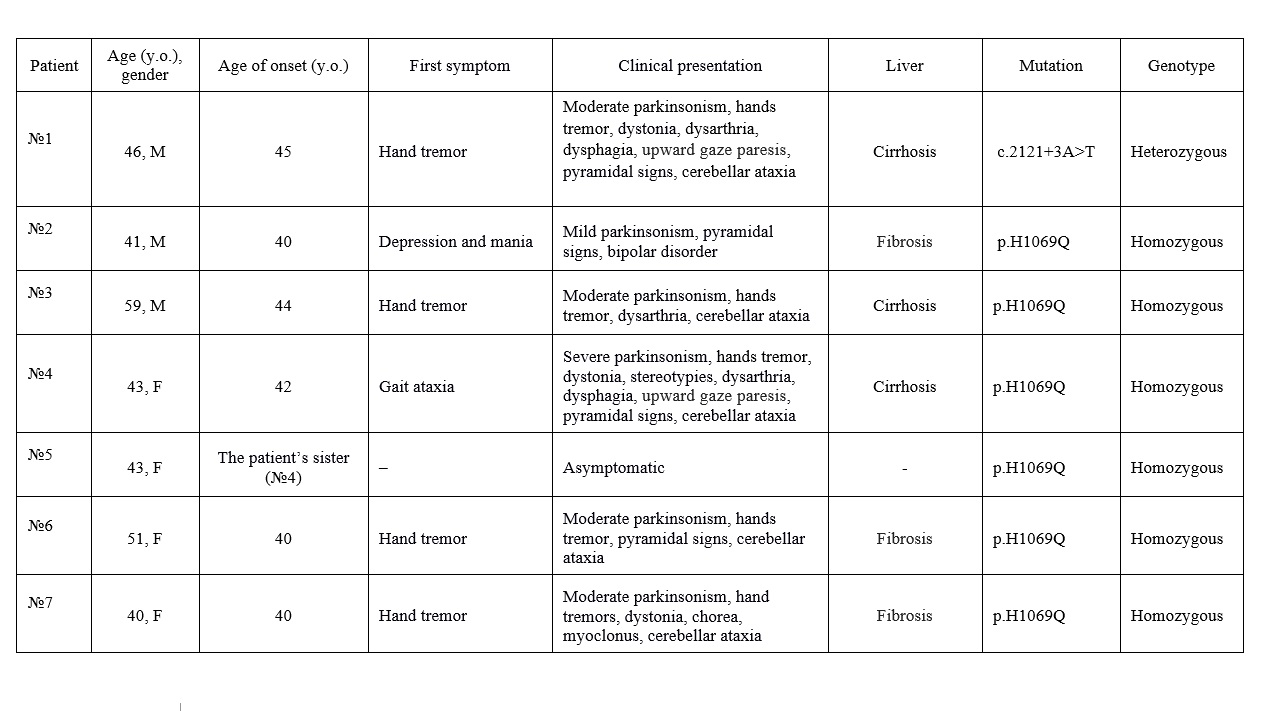
Characteristics of patients with late-onset WD
To cite this abstract in AMA style:
I. Minaev, E. Nuzhnyi, V. Poleschuk, A. Protopopova, N. Abramycheva, S. Illarioshkin. Late-onset neurologic Wilson’s disease: case series [abstract]. Mov Disord. 2024; 39 (suppl 1). https://www.mdsabstracts.org/abstract/late-onset-neurologic-wilsons-disease-case-series/. Accessed April 27, 2025.« Back to 2024 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/late-onset-neurologic-wilsons-disease-case-series/