Category: Rare Genetic and Metabolic Diseases
Objective: To report a novel variant of XK without the characteristic RBC phenotype
Background: XK disease (previously called McLeod Syndrome) is a rare X-linked disorder which presents with multisystemic manifestations. Patients who have the RBC McLeod phenotype but show minimal or no neurological features have been identified. We report the neurological presentation of a man with a novel missense variant of XK yet lacks the characteristic RBC phenotype.
Method: Case report
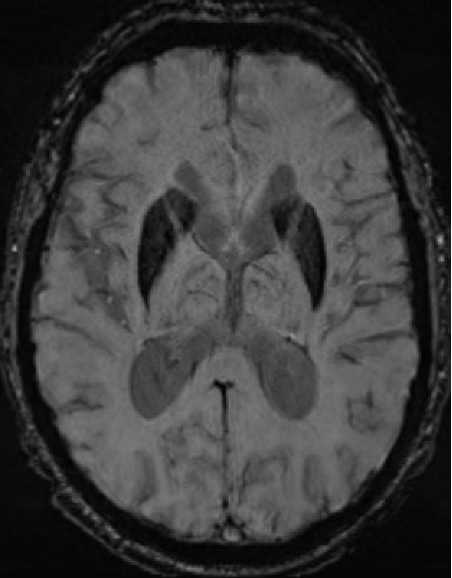
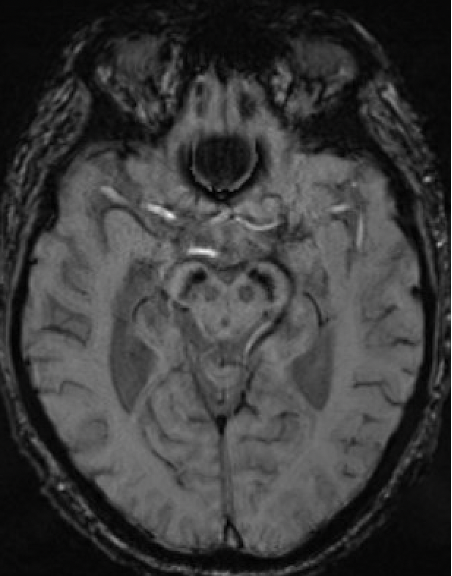
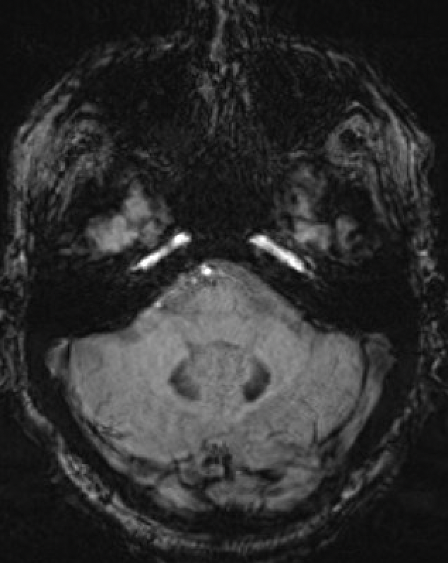
Results: A 67-year-old man presented with 11-year history of gradually progressive chorea. Cognitive screening showed impairment in working memory and executive function. The rest of the neurologic exam, including reflexes, was normal. At 70, chorea had generalized, even while on deutetrabenazine 24mg b.i.d. and aripiprazole 7.5mg q.d. At 73, MoCA was 8/30 with prominent frontal executive dysfunction, difficulty with visuospatial and memory domains. Throughout his disease course, he did not report any cardiac, autonomic, sensory, sleep or epileptic symptoms. Liver enzymes, serum copper, ceruloplasmin, iron, creatine kinase were normal. Serum ferritin was mildly elevated (832 ng/ml; Ref: 30-566). No acanthocytes were seen. ECG, echocardiography, EMG, and abdominal CT were unremarkable. Cranial MRI showed symmetric prominent hypointensity of the corpus striatum, substantia nigra and dentate nuclei with sparing of the thalami, most evident on the susceptibility weighted images [Figs 1-3]. Head CT did not show abnormal calcium deposition. Kell blood group system serologic typing of the RBCs revealed a K+k+, Kp(a-b+), Jsb+, Kx+ phenotype comparable to controls, not consistent with the McLeod phenotype. However, sequencing of the XK gene detected a novel missense c.121T>G change in exon 1 encoding a p.Leu41Val amino acid change predicted to be located in the 2nd transmembrane spanning region of the XK protein. Repeat expansion testing for the following genes was normal: HTT CAG (19/21 repeats), JPH3 (14/15), TBP (37/38), C9orf72 (2/2). No significant variants were found in: AP4M1, ATP13A2, C9orf12, COASY, CP, CRAT, DCAF17, FA2H, FTL, FUCA1, GJA1, GTPBP2, KIF1A, PANK2, PLA2G6, REPS1, SCP2, SLC25A42, SQSTM1, VPS13A, WDR45.
Conclusion: If confirmed through whole genome sequencing (ongoing analysis), this apparently contradictory situation may lead to insights into XK function and membrane trafficking in different organ systems.
To cite this abstract in AMA style:
J. Yu, R. Walker, A. Danek, C. Westhoff, S. Vege, I. Itin. Huntingtonism and bilateral basal ganglia hypointensity in a male with an XK variant but no McLeod red cell phenotype [abstract]. Mov Disord. 2023; 38 (suppl 1). https://www.mdsabstracts.org/abstract/huntingtonism-and-bilateral-basal-ganglia-hypointensity-in-a-male-with-an-xk-variant-but-no-mcleod-red-cell-phenotype/. Accessed December 12, 2025.« Back to 2023 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/huntingtonism-and-bilateral-basal-ganglia-hypointensity-in-a-male-with-an-xk-variant-but-no-mcleod-red-cell-phenotype/