Category: Rare Genetic and Metabolic Diseases
Objective: To analyze the pattern of movement disorder in a child with mitochondrial respiratory chain disorder
Background: Secondary movement disorders are often a clue to an underlying neurometabolic disorder in children. Mitochondrial disorders are a rare and under-recognized cause of movement disorders in children. They present with hyperkinetic disorders more commonly than hypokinetic disorders.
Method: An eight-year-old girl presented with frequent falls for the past 2 years, progressive speech abnormality for the past 8 months, and slowness in performing daily activities for the past 7 months. She also gradually developed action-induced intermitting twisting of limbs, stiffness, small and untidy handwriting, drooling of saliva, and excessive giggling. Past and family history was unremarkable. There was third-degree consanguinity in the family.
On examination, she was conscious and oriented, with mild cognitive impairment. She had a dull monotonous speech (extrapyramidal dysarthria), normal head circumference (51 cm), lower limb spasticity, generalized severe dystonia with bradykinesia, brisk deep tendon reflexes, bilateral extensor plantar and striatal toe. Her gait was abnormal with a mix of bradykinesia and spasticity. The clinical possibilities considered were neurodegeneration with brain iron accumulation, Wilson disease, Juvenile Huntington’s disease, and Leigh and Leigh-like syndromes.
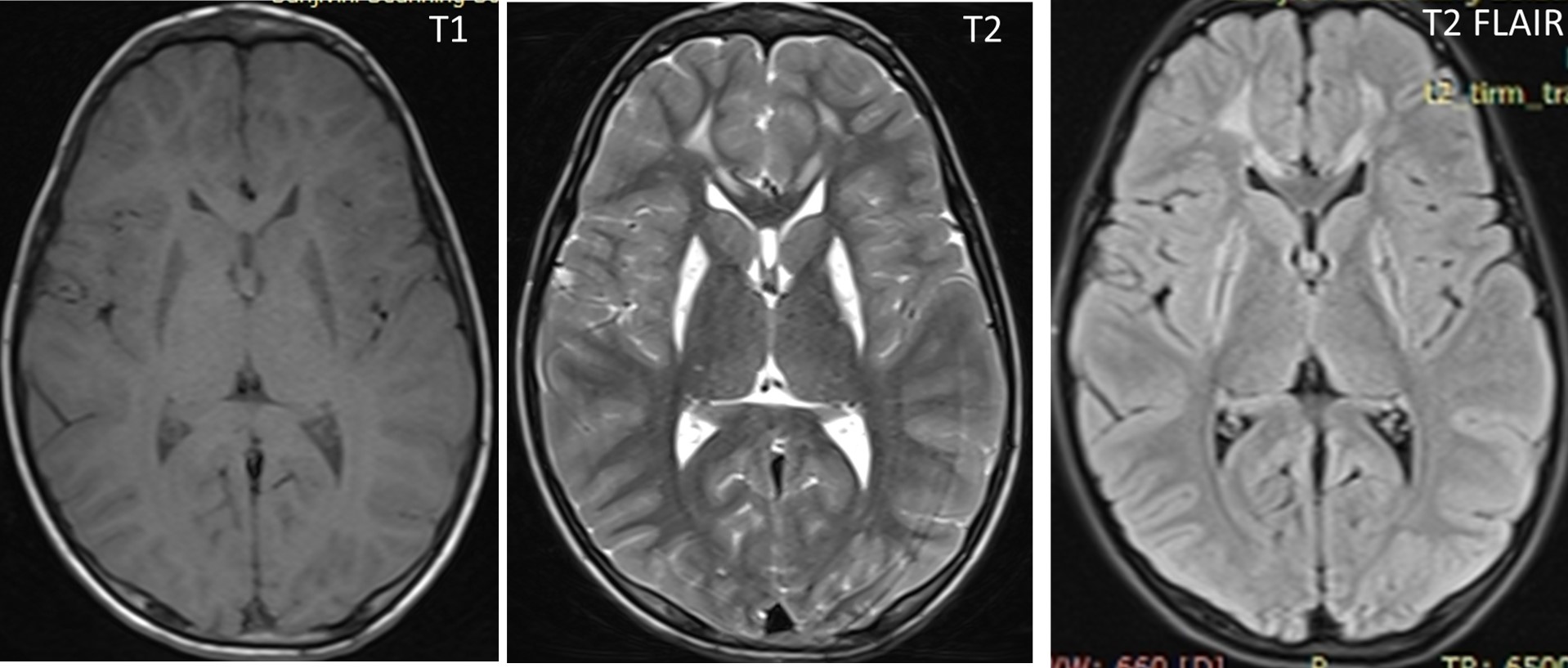
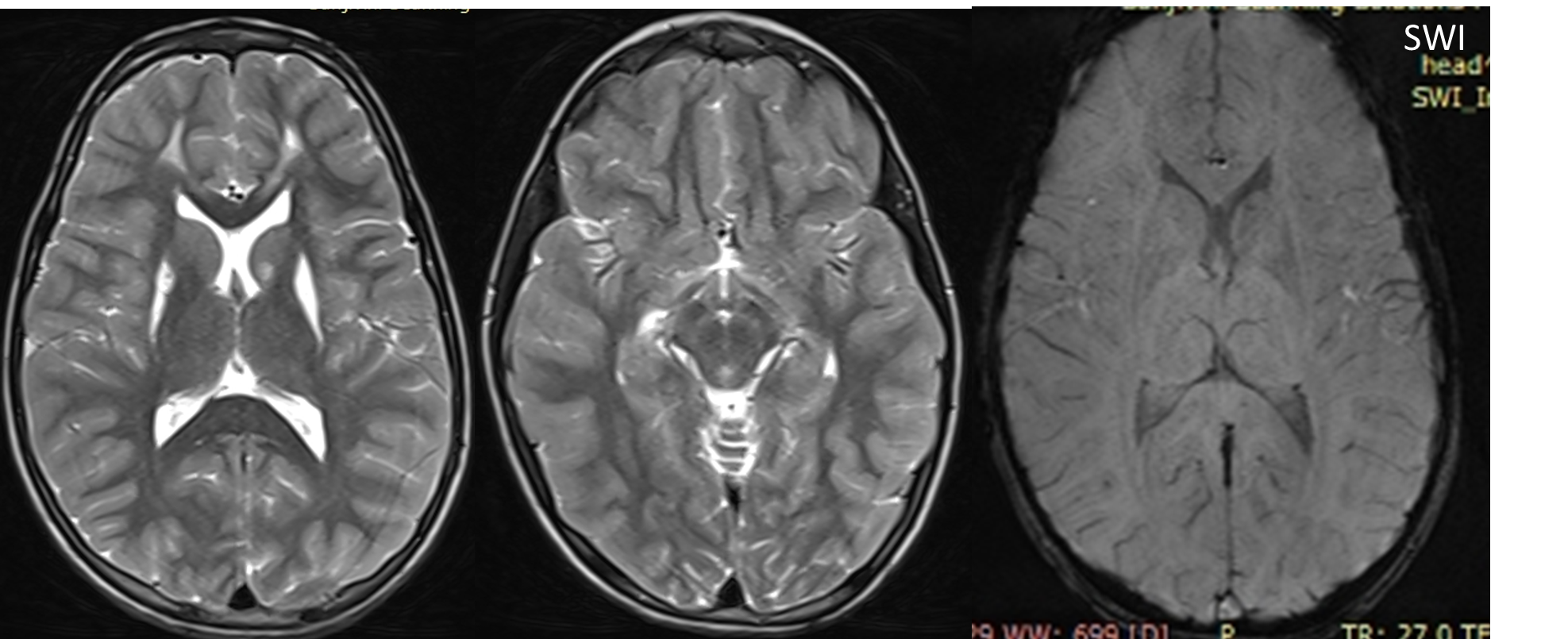
Results: Her MRI brain showed bilateral symmetrical hyperintensity in the putamina with cystic changes. There was bilateral hyperintensity in the periventricular white matter in the frontal area. Her liver and renal function tests, serum ceruloplasmin, urinary copper, and biotinidase enzyme levels were normal. There was mildly elevated arterial lactate and increased lactate in the urine on gas chromatography. The genetic analysis showed a pathogenic homozygous variant c.1268C>T (p.Thr423Met) on exon 9 in the NDUFV1 gene, confirming a diagnosis of autosomal recessive, nuclear-encoded complex 1 deficiency as a cause of late presentation of Leigh syndrome in the index case.
Conclusion: Complex I deficiency is a common cause of Leigh disease and late-onset forms rarely present with movement disorders. Nuclear-encoded autosomal recessive variants are more common than mitochondrial inheritance. The radiological clue is the involvement of bilateral basal ganglia lesions.
To cite this abstract in AMA style:
A. Saini, S. Khanna. Generalized dystonia and spasticity: The fault in the mitochondria [abstract]. Mov Disord. 2023; 38 (suppl 1). https://www.mdsabstracts.org/abstract/generalized-dystonia-and-spasticity-the-fault-in-the-mitochondria/. Accessed April 20, 2025.« Back to 2023 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/generalized-dystonia-and-spasticity-the-fault-in-the-mitochondria/