Session Information
Date: Sunday, October 7, 2018
Session Title: Dystonia
Session Time: 1:45pm-3:15pm
Location: Hall 3FG
Objective: To assess the frequency of KMT2B mutations in a cohort of patients with childhood-onset dystonia and characterize the related molecular and phenotypic spectrum.
Background: Heterozygous mutations in KMT2B, encoding a histone lysine methyltransferase, were recently reported as a cause of childhood-onset generalized dystonia variably associated with additional neurological and systemic features.
Methods: 55 patients (23F,22M) with genetically undefined childhood-onset dystonia were screened by means of Whole Exome Sequencing or customized gene panels including known dystonia-associated genes. KMT2B variants were confirmed by Sanger sequencing and further characterized by segregation analysis, bioinformatics tools and in silico homology modelling in some cases.
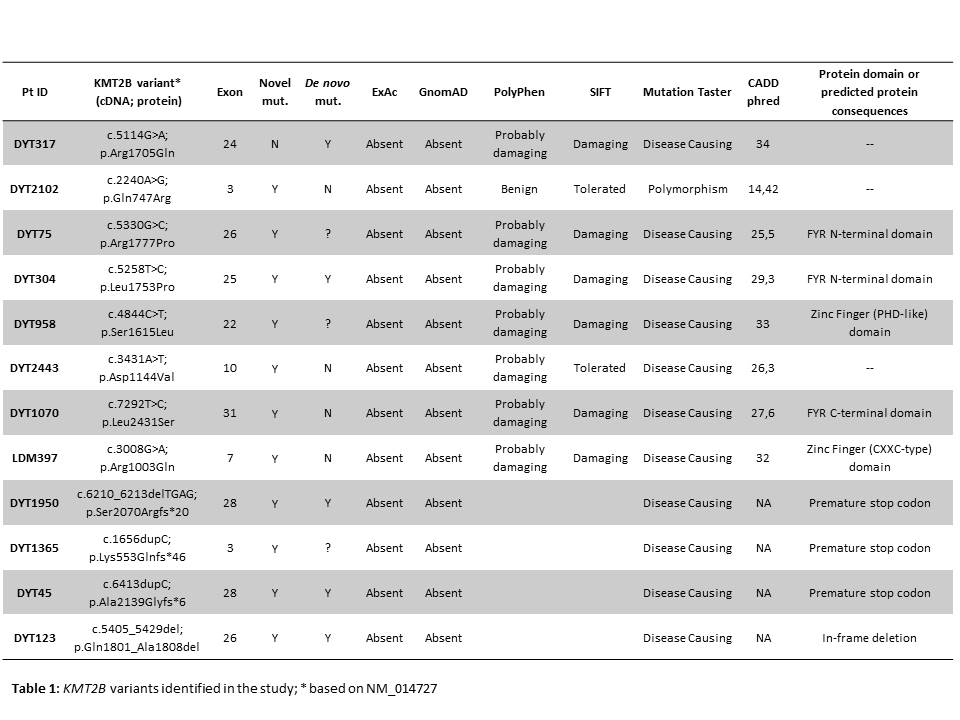
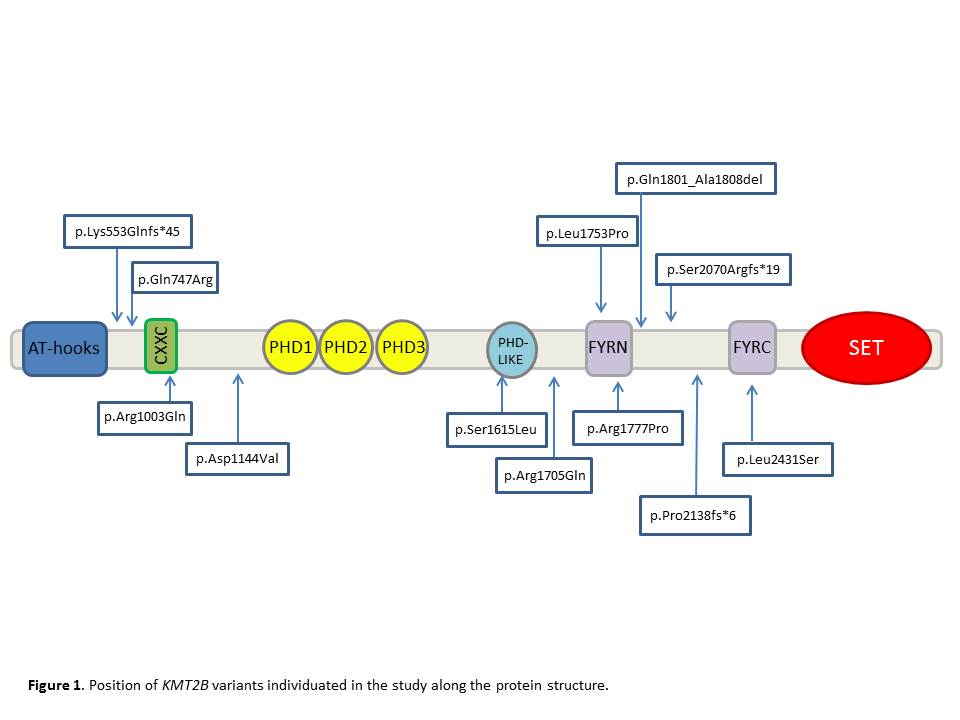
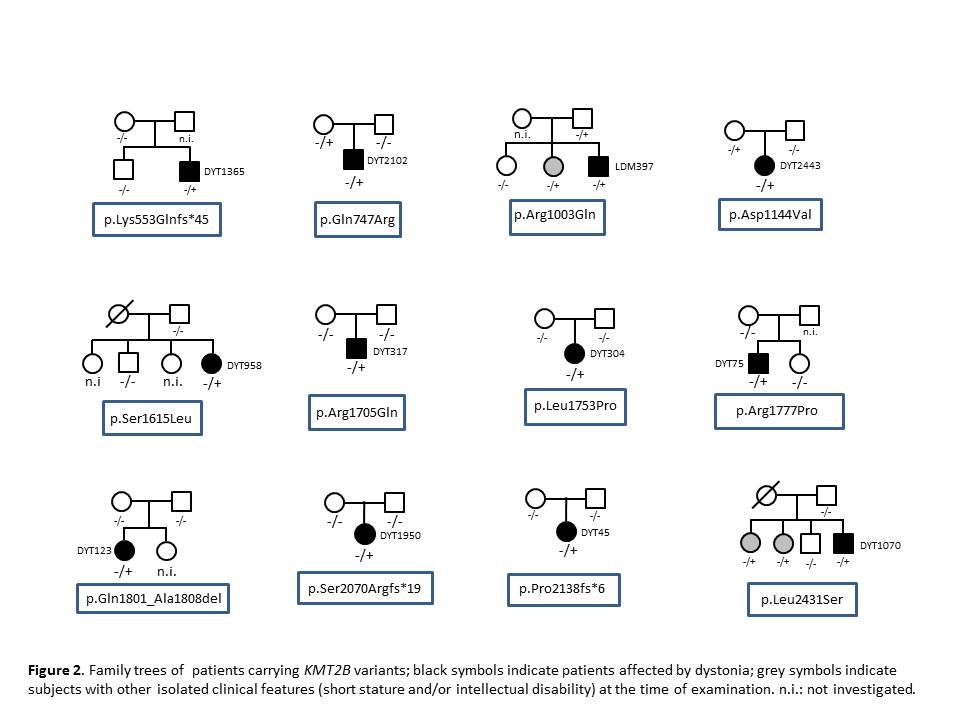
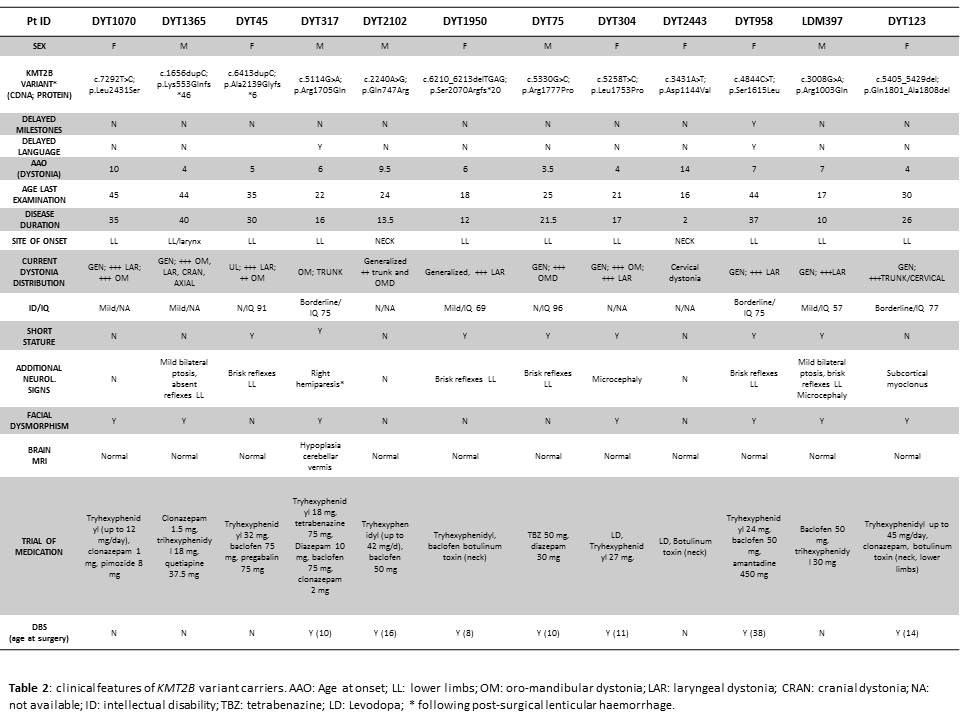
Results: 12/55 (21.8%) patients were found to carry KMT2B variants (8 missense, 3 frameshift, 1 in-frame deletion) [table 1] [figure 1], of which 5 were confirmed to be de novo, 4 were inherited from one unaffected parent and 3 were likely de novo segregating with disease status in available family members [figure 2]. All but one variant had not been reported previously. Mean age at onset was 6.5 years (range 3.5-14); mean age at last examination was 28 years (range 17-45) and mean disease duration was 22.5 years (range 2-40). 10/12 (83%) patients had lower limb presentation, 11/12 (92%) progressed to generalization with laryngeal dystonia observed in 10/12 (83%). Mild intellectual disability, short stature, minor facial dysmorphisms were recurrently observed [table 2]. Brain MRI was normal in all cases, including DWI/SWI sequences. 7/12 patients underwent bilateral pallidal DBS with excellent, long-lasting motor improvement (mean post-operative follow-up: 12 years). Additional genetic findings included a novel, de novo GNAO1 missense mutation (p.Cys215Tyr) identified in a 42-year-old male with a 30-year history of myoclonus dystonia and biallelic ATM mutations (p.Cys907*/p.Glu2052Lys) in a 40-year-old female with isolated generalized dystonia presenting at age 13.
Conclusions: Mutations in KMT2B are a relevant cause of dystonia in the paediatric population, with autosomal dominant inheritance and possible incomplete penetrance in some cases. Red flags include onset in the lower limbs, caudo-cranial spreading, oro-mandibular and laryngeal involvement, short stature and mild intellectual disability. Brain MRI in our cases did not confirm previously reported basal ganglia alterations. Childhood-onset dystonia can rarely be an atypical presentation of other genetic diseases.
References: Meyer E, Carss KJ, Rankin J, et al. Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat Genet 2017;49:223-237. Zech M, Boesch S, Maier EM, et al. Haploinsufficiency of KMT2B, encoding the lysine-specific histone methyltransferase 2b, results in early-onset generalized dystonia. Am J Hum Genet 2016;99:1377-1387.
To cite this abstract in AMA style:
M. Carecchio, G. Zorzi, F. Invernizzi, C. Panteghini, L. Romito, F. Zibordi, V. Leuzzi, S. Galosi, P. Morana, B. Morana, C. Piano, A. Bentivoglio, C. Reale, F. Girotti, M. Topf, A. Joseph, M. Kurian, S. Lubbe, B. Garavaglia, N. Mencacci, N. Nardocci. Frequency and Phenoptypic Spectrum of KMT2B Mutations in Childhood-Onset Dystonia: Results from a Single-Centre Cohort Study [abstract]. Mov Disord. 2018; 33 (suppl 2). https://www.mdsabstracts.org/abstract/frequency-and-phenoptypic-spectrum-of-kmt2b-mutations-in-childhood-onset-dystonia-results-from-a-single-centre-cohort-study/. Accessed May 6, 2026.« Back to 2018 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/frequency-and-phenoptypic-spectrum-of-kmt2b-mutations-in-childhood-onset-dystonia-results-from-a-single-centre-cohort-study/