Objective: To identify the genetic cause of disease in a French family with multiple members affected by dystonic tremor with autosomal dominant inheritance.
Background: Spinocerebellar ataxia 21 (SCA21) is a slowly progressive early-onset autosomal dominant cerebellar ataxia variably combined with cognitive impairment, parkinsonism, dystonia, and myoclonus. Pathogenic variants in the TMEM240 gene are the cause of SCA21. Around 60 cases of TMEM240-related disease were reported to date, with the P170L variant recurring in several families with independent origin and different clinical phenotype: the most common manifestations are ataxia and cognitive impairment though in some cases dystonia has been reported as presenting feature. 1–10
Method: Seven subjects were clinically evaluated by neurologists with additional training in movement disorders and a whole-exome sequencing (WES) was performed. The candidate variants were confirmed through Sanger sequencing.
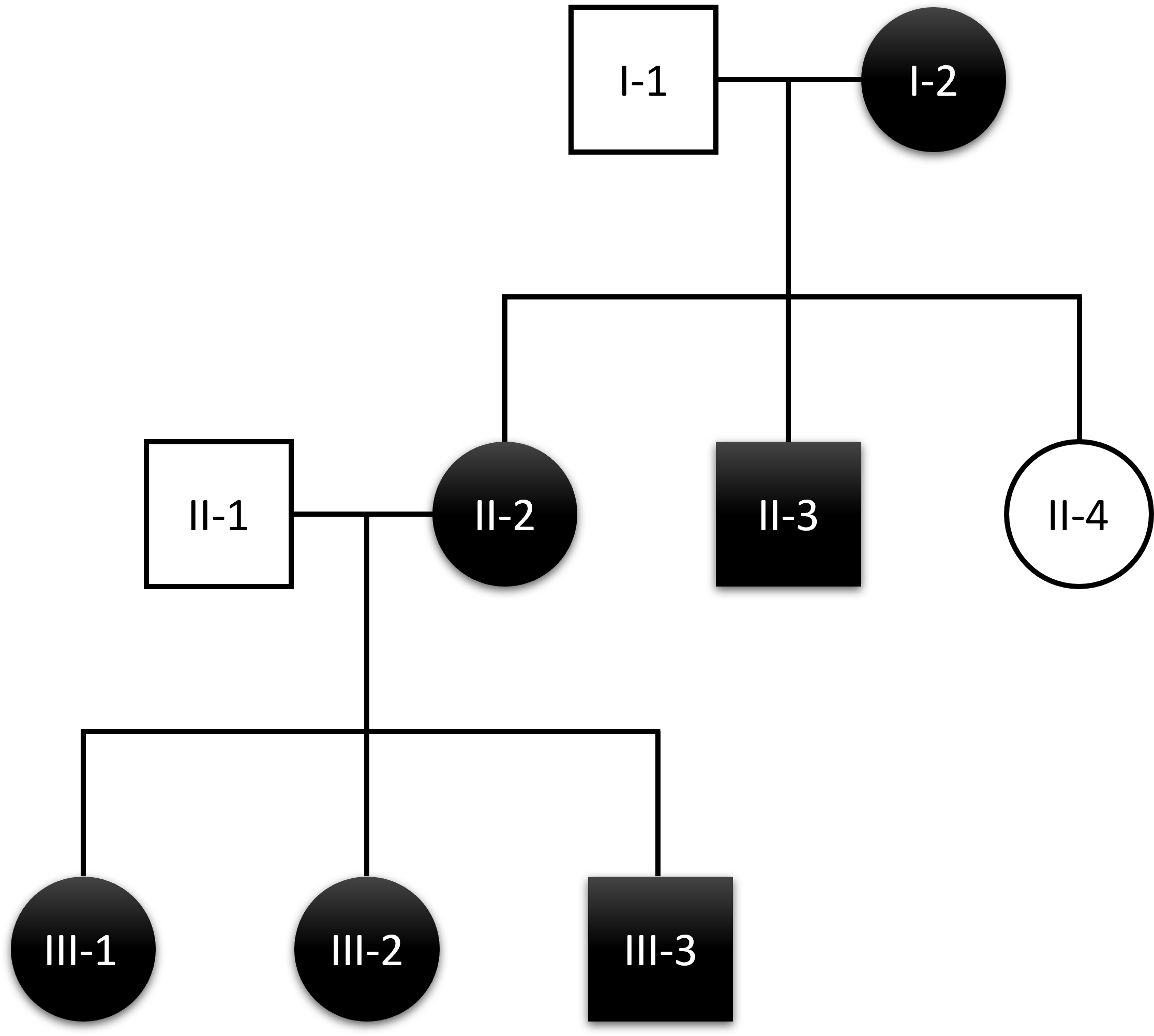
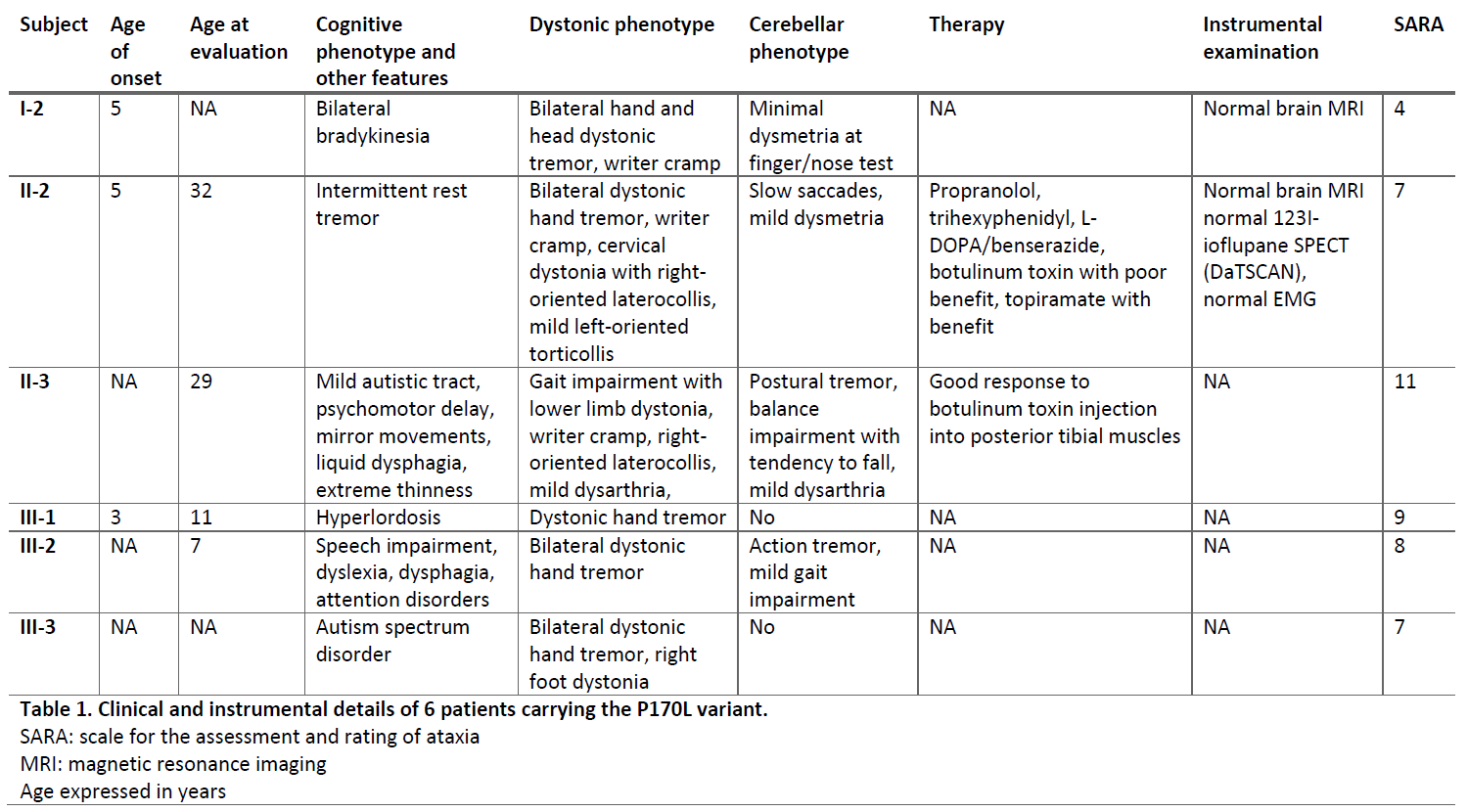
Results: Six subjects, four female and two male, belonging to three consecutive generations, presented an autosomal dominant tremulous dystonia with disease onset at 4-6 years. They displayed dystonic tremor of upper and lower limbs, neck, and head, as well as writer’s cramp and foot dystonia. Additional neurological signs interested some subjects including minimal dysmetria at finger-nose test, action tremor, bradykinesia, saccade impairment, dysphagia, autism spectrum disorder, and learning disability. All of them presented the recurrent P170L TMEM240 variant at WES and at Sanger sequencing confirmation. All instrumental examinations including brain MRI, DaTscan, and EMG resulted normal. One additional subject complained hand tremor by the age of 12 years without dystonia nor ataxia. The TMEM240 pathogenic variant was not present in this subject, thus she likely represents a phenocopy.
Conclusion: This work expands the clinical spectrum of TMEM240-related disease, including dystonic tremor as a main clinical presentation.
References: 1. Delplanque, J. et al. TMEM240 mutations cause spinocerebellar ataxia 21 with mental retardation and severe cognitive impairment. Brain 137, 2657–2663 (2014).
2. Ward, J. M. & la Spada, A. R. Identification of the SCA21 disease gene: remaining challenges and promising opportunities. Brain 137, 2626–2628 (2014).
3. Zeng, S. et al. Spinocerebellar ataxia type 21 exists in the Chinese Han population. Sci Rep 6, 3–6 (2016).
4. Yahikozawa, H. et al. A Japanese Family of Spinocerebellar Ataxia Type 21: Clinical and Neuropathological Studies. Cerebellum 17, 525–530 (2018).
5. Traschütz, A. et al. The movement disorder spectrum of SCA21 (ATX-TMEM240): 3 novel families and systematic review of the literature. Parkinsonism Relat Disord 62, 215–220 (2019).
6. Burdekin, E. D. et al. The Neurodevelopmental and Motor Phenotype of SCA21 (ATX-TMEM240). J Child Neurol 35, 953–962 (2020).
7. Camargo, C. H. F., Piva-Silva, A. K. C., Munhoz, R. P., Raskin, S. & Teive, H. A. G. Spinocerebellar ataxia type 21 (TMEM240) with tremor and dystonia. Eur J Neurol 28, e63–e64 (2021).
8. Cherian, A., Divya, K. P., Vijayaraghavan, A. & Krishnan, S. Pearls & Oy-sters: SCA21 Due to TMEM240 Variation Presenting as Myoclonus Dystonia Syndrome. Neurology 99, 531–534 (2022).
9. van der Put, J. et al. On Spinocerebellar Ataxia 21 as a Mimicker of Cerebral Palsy. Neurol Genet 8, 1–5 (2022).
10. Park, D. G., Kim, M. S. & Yoon, J. H. The First Korean Family of Spinocerebellar Ataxia 21 (ATX-TMEM240) with Facial Dystonic Phenotype. Cerebellum 21, 6–8 (2022).
To cite this abstract in AMA style:
V. Yahya, E. Monfrini, E. Moro, A. Di Fonzo. Dystonic tremor as main manifestation of a large SCA21 family [abstract]. Mov Disord. 2023; 38 (suppl 1). https://www.mdsabstracts.org/abstract/dystonic-tremor-as-main-manifestation-of-a-large-sca21-family/. Accessed March 26, 2026.« Back to 2023 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/dystonic-tremor-as-main-manifestation-of-a-large-sca21-family/