Category: Parkinson's Disease: Genetics
Objective: To describe the phenotype of three siblings with early-onset parkinsonism and epilepsy due to an x-linked phosphoglycerate kinase 1 deficiency, expanding our understanding of this rare mutation.
Background: X-linked parkinsonism encompasses rare heterogeneous disorders, being more prevalent in males. The phosphoglycerate kinase 1 (PGK1, OMIM 300653) deficiency was first described in 1968 as a cause of non-spherocytic hemolytic anemia (1). It is a recessive metabolic disease, and the clinical picture that occurs in men involves erythrocytes, skeletal muscle, and the central nervous system (2). A rare presentation similar to Parkinson’s disease (PD) can also occur, previously described in only eight cases in the literature (3).
Method: A case report of three male brothers with an x-linked PGK1 deficiency with early-onset parkinsonism and epilepsy.
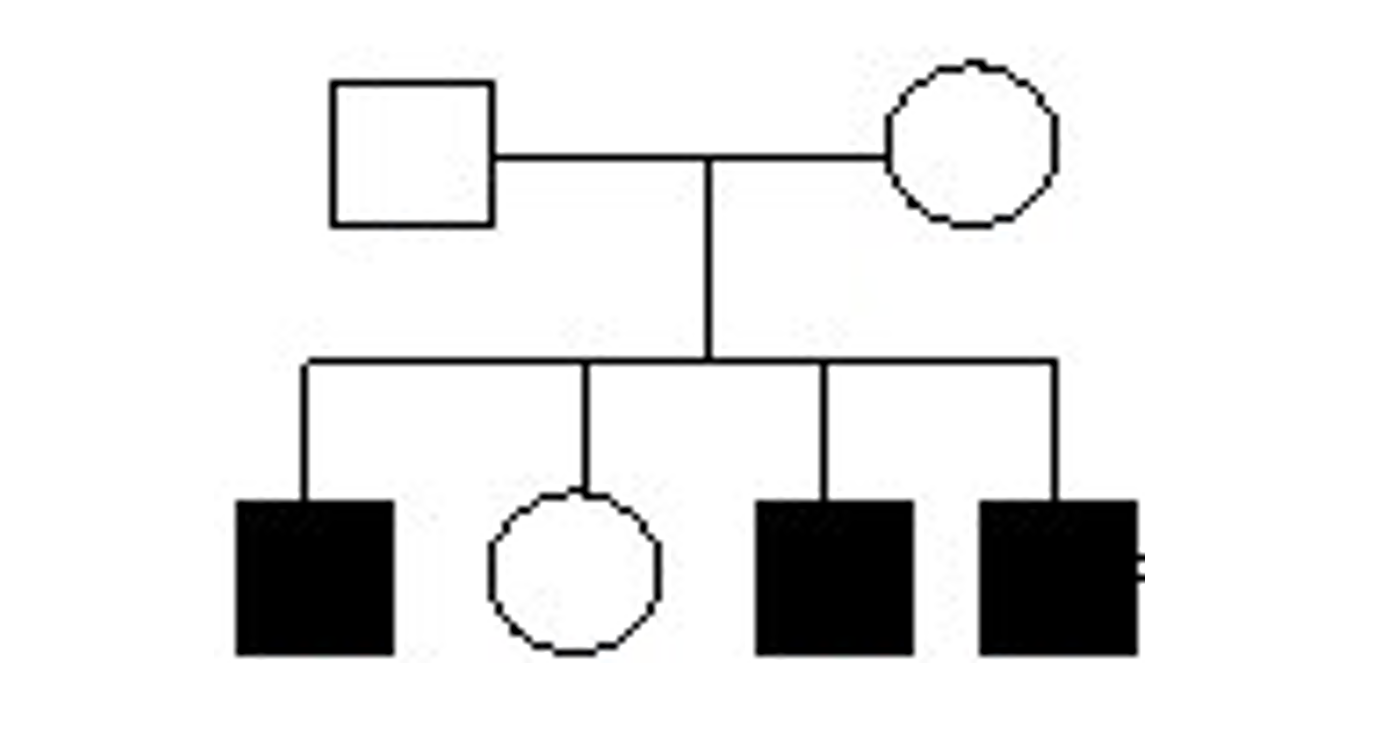
Results: Three male siblings born to non-consanguineous healthy parents presented with parkinsonism at 23, 25, and 27 years old. They had concomitant cognitive complaints, neuropsychiatric features (hallucinations and delusions), and generalized epilepsy. The seizures started in early childhood and were easily controlled with carbamazepine. There was a healthy sister (figure 1). At the examination, all three patients had asymmetric akinetic-rigid parkinsonism, two presented action tremor, and one had intermittent dystonic postures in the lower limb (figure 2). They demonstrated excellent response to levodopa, with mild wearing-off and no dyskinesia. Their psychotic symptoms were well controlled with quetiapine and worsened with dopaminergic agonists. On investigation, MRI and laboratory findings were unremarkable. Electroencephalogram showed occasional frontocentral paroxysms. The Next-Generation Sequencing panel for hereditary parkinsonism, including epilepsy-associated genes (DNAJC6, SYNJ1, RAB39B), was negative. An exome was requested, demonstrating a pathogenic hemizygous variant in the PGK1 gene (p.Gly317Asp), absent in the population database and predicted in silico to be deleterious.
Conclusion: Among adult-onset x-linked hereditary parkinsonism with a phenotype similar to PD and concomitant epilepsy, these cases illustrate that we should consider PGK1 mutation. Therefore, this concept might guide molecular testing to a more robust case series investigating its natural history and, in the future, specific treatments.

References: 1. Kraus AP, Langston MF, Lynch BL. Red cell phosphoglycerate kinase deficiency. A new cause of non-spherocytic hemolytic anemia. Biochem Biophys Res Commun 1968;30(2):173–177.
2. Di Lazzaro G, Magrinelli F, Estevez-Fraga C, Valente EM, Pisani A, Bhatia KP. X-Linked Parkinsonism: Phenotypic and Genetic Heterogeneity. Mov Disord. 2021 Jul;36(7):1511-1525.
3. Morales-Briceño H, Ha AD, London K, Farlow D, Chang FCF, Fung VSC. Parkinsonism in PGK1 deficiency implicates the glycolytic pathway in nigrostriatal dysfunction. Parkinsonism Relat Disord 2019;64:319–323.
To cite this abstract in AMA style:
J. Parmera, T. Guimarães, R. Cury, F. Freua, E. Barbosa, F. Kok. Dopamine-responsive x-linked parkinsonism-epilepsy due to phosphoglycerate kinase-1 deficiency [abstract]. Mov Disord. 2023; 38 (suppl 1). https://www.mdsabstracts.org/abstract/dopamine-responsive-x-linked-parkinsonism-epilepsy-due-to-phosphoglycerate-kinase-1-deficiency/. Accessed April 26, 2026.« Back to 2023 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/dopamine-responsive-x-linked-parkinsonism-epilepsy-due-to-phosphoglycerate-kinase-1-deficiency/