Session Information
Date: Saturday, October 6, 2018
Session Title: Genetics (Non-PD)
Session Time: 1:45pm-3:15pm
Location: Hall 3FG
Objective: To determine the causal locus and functional mechanism associated with XDP using integrative genomics methods in patient-derived and genome-edited induced pluripotent stem cell (iPSC) models.
Background: XDP is a neurodegenerative movement disorder with variable clinical presentation. The genetic cause has been attributed to seven disease specific changes (DSCs) observed in all cases on Xq13.1. None of the DSCs have annotated functions and previous studies have suggested the region is recalcitrant to recombination. The causal mechanism has thus remained elusive.
Methods: We established an international consortium for XDP and aggregated specimens from 792 individuals (403 XDP males, 23 heterozygous carrier females, 352 controls, and 14 non-manifesting carriers). We developed fibroblast lines (n=45) and an iPSC resource from 12 patients and their unaffected relatives, including differentiation to four neuronal lineages. We then integrated genome and transcriptome assembly using six technologies (Illumina whole-genome sequencing [WGS]; 10X Genomics assembly; DNA capture sequencing [CapSeq]; RNA CapSeq; Pacific Biosciences long-read RNA CapSeq; total RNAseq).
Results: Genomic analyses detected all 7 DSCs and 47 additional XDP-associated variants. Haplotype reconstruction further revealed five recombination events and eight derivatives of the most common haplotype in this cohort, suggesting that recombinations occur regularly in XDP, and narrowing the critical locus to a region that includes TAF1 exclusively. Transcriptome assembly and expression analyses discovered a signature associated with XDP in TAF1 that involved aberrant splicing and intron retention (IR) that terminated in proximity to an intron 32 SVA insertion and negatively correlated with TAF1 expression in XDP probands. The hexameric repeat in this SVA was found to be polymorphic in probands and unstable in culture, ranging from 35-52 repeats, and inversely correlated with age at onset of XDP (r2=0.5). Remarkably, CRISPR/Cas9 excision of the SVA rescued the aberrant splicing signatures and normalized overall TAF1 expression in XDP cells [figure1]. Transcriptome profiling across diverse neuronal lineages is ongoing; analyses to date suggests convergence on multiple neurodevelopmental and neurodegenerative pathways and co-expression networks.
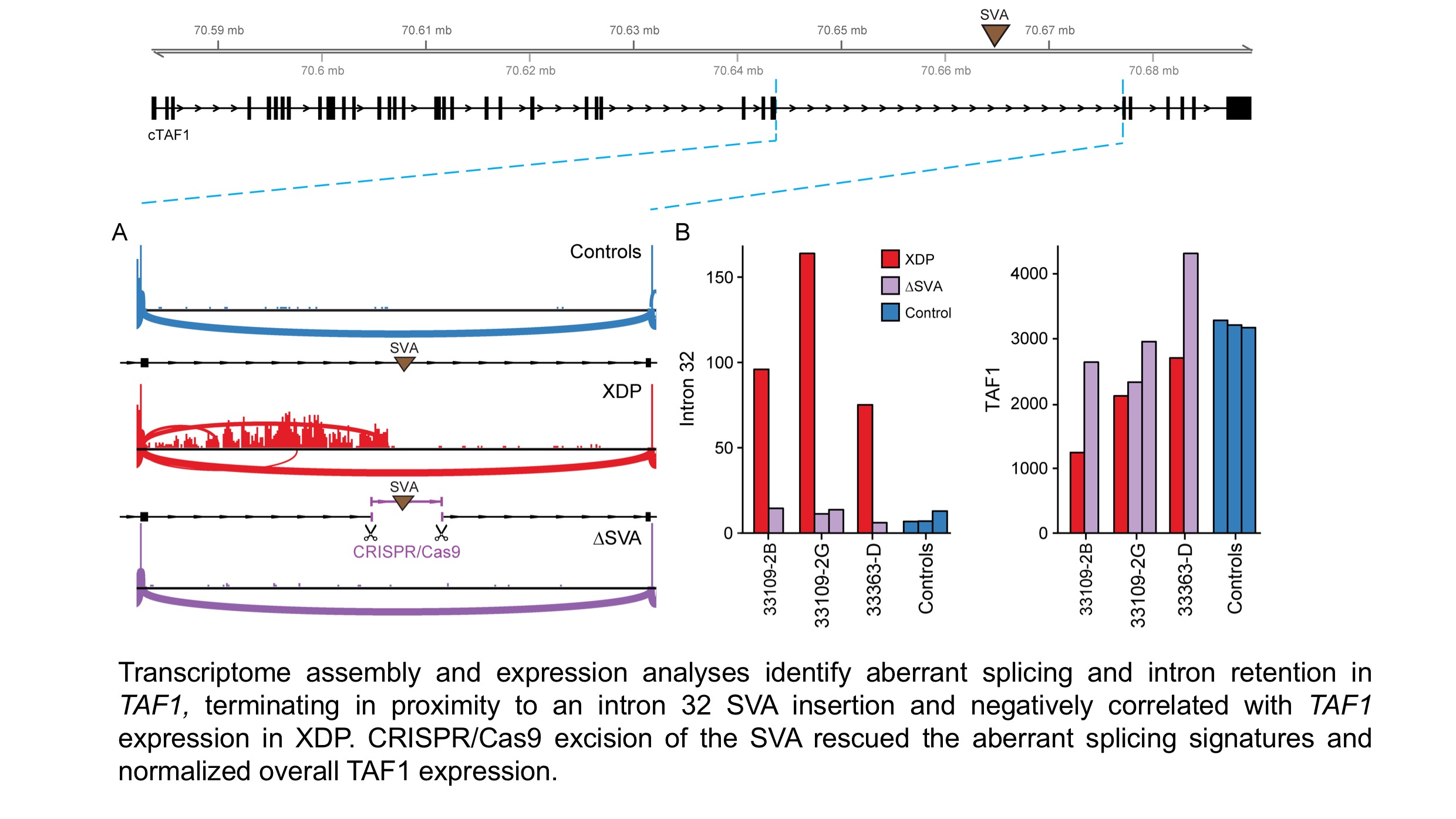
Conclusions: These studies suggest a novel transcriptional signature associated with an SVA insertion in XDP. Disease onset correlated with SVA repeat length, and aberrant transcriptional signatures could be rescued using CRISPR/Cas9 editing, suggesting a potential therapeutic target.
To cite this abstract in AMA style:
A. Domingo, S. Sharma, A. Aneichyk, W. Hendriks, Y. Yadav, D. Shin, D. Gao, C. Vaine, B. Currall, N. Kulkarni, T. Multhaupt-Buell, E. Penney, M. Dy, C. Go, RD. Jamora, R. Rosales, M. Ang, U. Müller, C. Klein, P. Acuña, X. Breakefield, L. Ozelius, DC. Bragg, M. Talkowski. Dissecting molecular signatures of X-linked dystonia-parkinsonism (XDP) through integrative genomics studies [abstract]. Mov Disord. 2018; 33 (suppl 2). https://www.mdsabstracts.org/abstract/dissecting-molecular-signatures-of-x-linked-dystonia-parkinsonism-xdp-through-integrative-genomics-studies/. Accessed July 4, 2026.« Back to 2018 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/dissecting-molecular-signatures-of-x-linked-dystonia-parkinsonism-xdp-through-integrative-genomics-studies/