Category: Rare Genetic and Metabolic Diseases
Objective: To report on the clinical spectrum of variants affecting dehydrodolichol diphosphate synthetase (DHDDS) and nuclear undecaprenyl pyrophosphate Synthase 1 (NUS1) and particularly to highlight their phenotypic overlap in line with their shared role in dolichol synthesis. We aim to inform movement disorder neurologists of the key clinical features which should prompt consideration of DHDDS/NUS1 related disease.
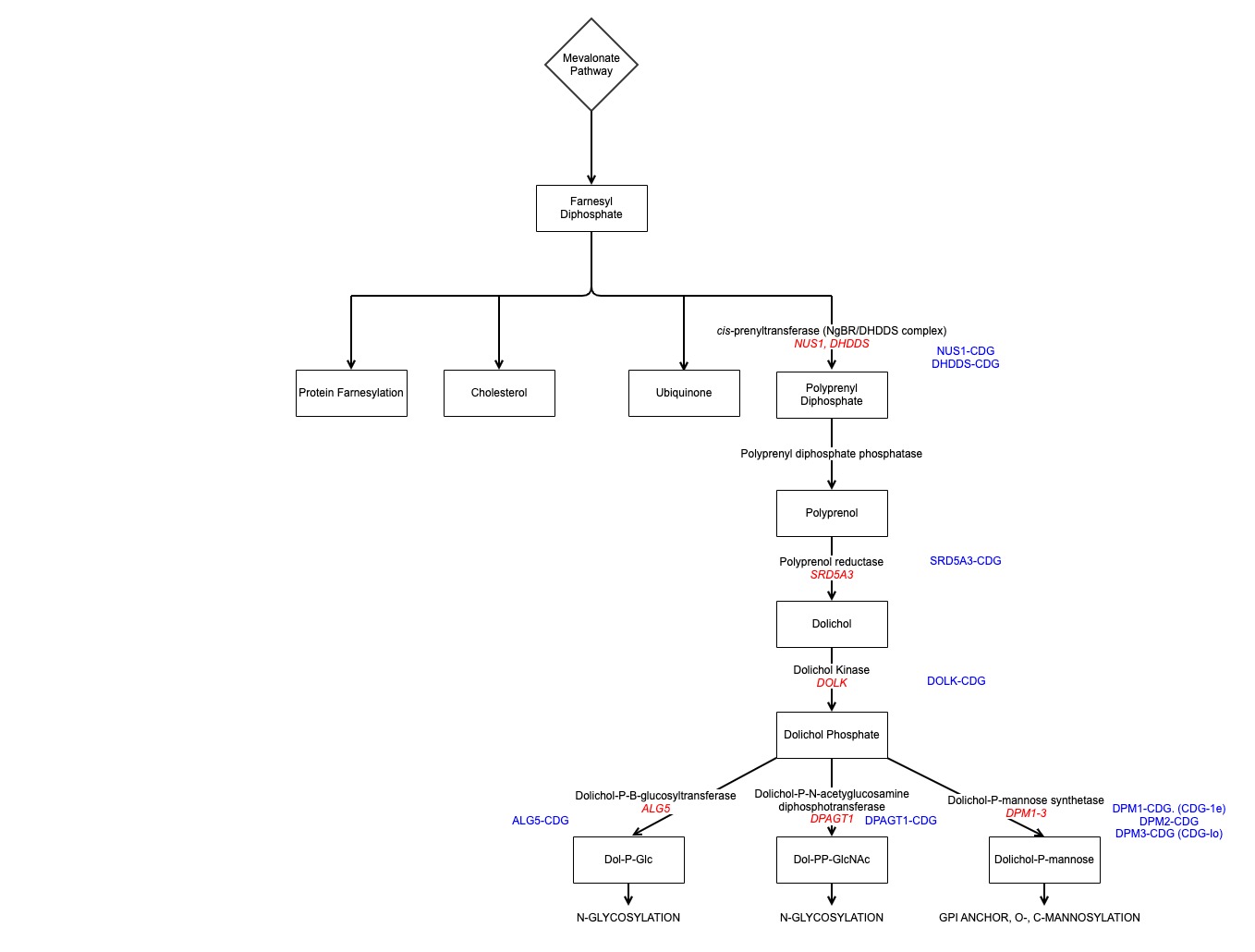
Background: The primary congenital disorders of glycosylation (CDGs) are a group of metabolic disorders with varied phenotypes. We present seven patients with remarkably similar phenotype, due to defects in the dolichol phosphate synthesis pathway, a lipid carrier needed in N-glycosylation, O-mannosylation, C-mannosylation and glycosylphosphatidylinositol anchor formation 1.
All harbour genetic mutations affecting the endoplasmic reticulum based cis-prenyltransferase (cis-PTase), an enzyme essential in early development. Normal cis-PTase acts at an early stage of dolichol synthesis and requires interaction of Nogo-B receptor (NgBr) and DHDDS subunits, encoded by NUS1 and DHDDS respectively. Such patients have been reported as progressive myoclonic ataxia or developmental and epileptic encephalopathy. Detailed accounts of associated movement disorders infrequent 2.
Method: Through multicentre collaboration we recruited seven patients with variants in DHDDS (n=3) and NUS1 (n=4). We present video examination and detailed descriptions. We review the literature to complement our phenotypic descriptions.
Results: A strikingly convergent phenotype of developmental delay, myoclonus, and ataxia, with and without a seizure disorder, was seen. Infantile and childhood onset seizures varied in semiology. Fever sensitivity was common. The complex movement disorder spanned myoclonus, ataxia, tremor, chorea, dystonia and parkinsonism as a later feature in some. Facial myoclonus, dysmorphic features and transient episodic deterioration are notable features. Patients may remain stable for decades. Normal investigations, including normal transferrin isoform profile, may falsely steer the clinician away from consideration of CDGs.
Conclusion: These patients were all encountered in movement disorder clinics. Previous literature has frequently dwelled on the epileptic phenotype. Global developmental delay, (frequently mild) epilepsy, action-induced myoclonus, insidious ataxia, and later onset parkinsonism are clinical clues.
References: 1. Edani B, Grabińska K, Zhang R, et al. Structural elucidation of the cis-prenyltransferase NgBR/DHDDS complex reveals insights in regulation of protein glycosylation. Proceedings of the National Academy of Sciences of the United States of America 2020; 117: 20794–20802.
2. Galosi S, Edani B, Martinelli S, et al. De novo DHDDS variants cause a neurodevelopmental and neurodegenerative disorder with myoclonus. Brain : a journal of neurology 2021; 139: 16–17.
To cite this abstract in AMA style:
L. Williams, J. Qiu, S. Waller, N. Elserafy, M. Tchan, P. Procopis, H. Sampaio, S. Mohammad, H. Morales-Briceño, V. Fung. DHDDS and NUS1: A converging pathway and common phenotype [abstract]. Mov Disord. 2022; 37 (suppl 2). https://www.mdsabstracts.org/abstract/dhdds-and-nus1-a-converging-pathway-and-common-phenotype/. Accessed May 15, 2026.« Back to 2022 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/dhdds-and-nus1-a-converging-pathway-and-common-phenotype/