Session Information
Date: Monday, September 23, 2019
Session Title: Rare Genetic and Metabolic Diseases
Session Time: 1:45pm-3:15pm
Location: Les Muses Terrace, Level 3
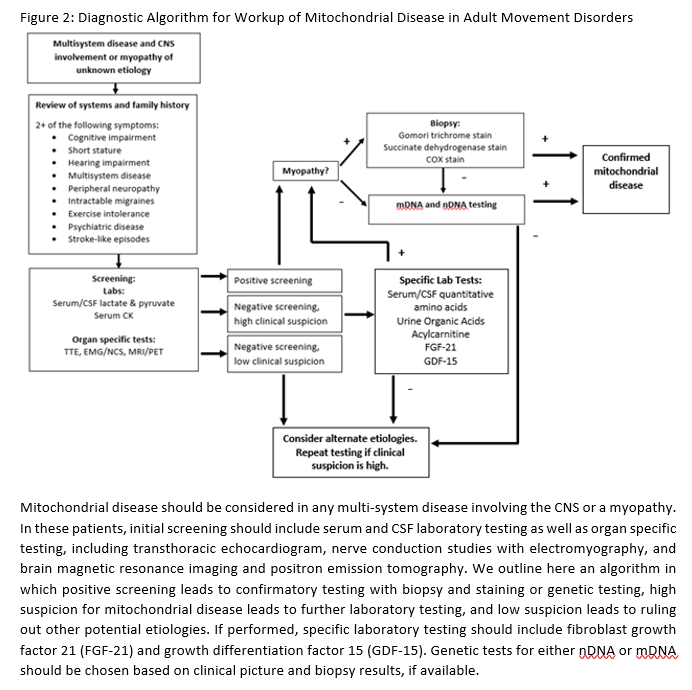
Objective: To present an algorithmic approach to the diagnosis of mitochondrial disease for clinical neurologists
Background: Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes (MELAS Syndrome) is a mitochondrial disease with a heterogeneous array of clinical and radiographic presentations which often mimic other neurologic diseases, rendering it challenging to diagnose.
Method: We present the cases of three members of a family, each with the same MELAS mutation. Patient 1 was diagnosed first, followed by two family members. One member was already being followed for presumed Parkinson’s disease; the other for migraines. Informed consent was obtained from all patients.
Results: Patient 1 presented with the gradual onset of worsening headaches, confusion, memory loss, difficulty reading and writing, dizziness, and syncope. She was found to have left temporal hyperintensities on brain MRI, thought suspicious for neoplasm. Serum lactate was initially normal but subsequently elevated. CSF lactate was also elevated. Acylcarnitine labs showed mildly elevated C6-DC. Genetic testing confirmed a heteroplasmic m.3243A>G mutation in MTTL1. Patient 2 had intractable migraines and was found to have normal CK, MRI, TTE, and NCS/EMG. Serum lactate was elevated only during acute pancreatitis episodes. MELAS was confirmed with genetic testing. Patient 3 carried a diagnosis of Parkinson’s disease and also had no abnormalities on MRI, TTE, or NCS/EMG. Patient 3 has not undergone confirmatory genetic testing but continues to be followed in clinic for management of presumed MELAS. Pedigree and clinical details are given in Figure 1. [figure1]
Conclusion: MELAS should be considered in patients presenting with multisystem disease, particularly when involving the CNS or a myopathy. If either a review of systems or family history is suggestive of mitochondrial disease, initial screening serum and CSF laboratory tests as well as organ-specific tests, must be obtained. If clinical suspicion is high, more specific laboratory testing should follow. Biopsy/staining or genetic testing are confirmatory. Through an algorithmic approach (Figure 2), a clinician can diagnose mitochondrial disease in a sensitive and efficient manner. [figure2]

To cite this abstract in AMA style:
N. Pulley, C. Condon, I. Haq. Detecting unsuspected mitochondrial disease: an algorithmic approach [abstract]. Mov Disord. 2019; 34 (suppl 2). https://www.mdsabstracts.org/abstract/detecting-unsuspected-mitochondrial-disease-an-algorithmic-approach/. Accessed April 25, 2025.« Back to 2019 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/detecting-unsuspected-mitochondrial-disease-an-algorithmic-approach/