Category: Ataxia
Objective: Provide insight into SACS mutations in India, by targeted gene panel of a suspected cohort.
Background: Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS), caused by mutations in SACS gene, consists of a triad of cerebellar ataxia, lower extremity spasticity and peripheral neuropathy. [1,2]
Method: Patients with 3 cardinal features of ARSACS were included. 9 were clinically identified from 215 cerebellar ataxia patients and evaluated genetically. Additional investigations included magnetic resonance imaging (MRI), fundoscopy, optical coherence tomography (OCT), nerve conduction studies and functional disability assessment by Spinocerebellar Degeneration Functional Score (SDFS).
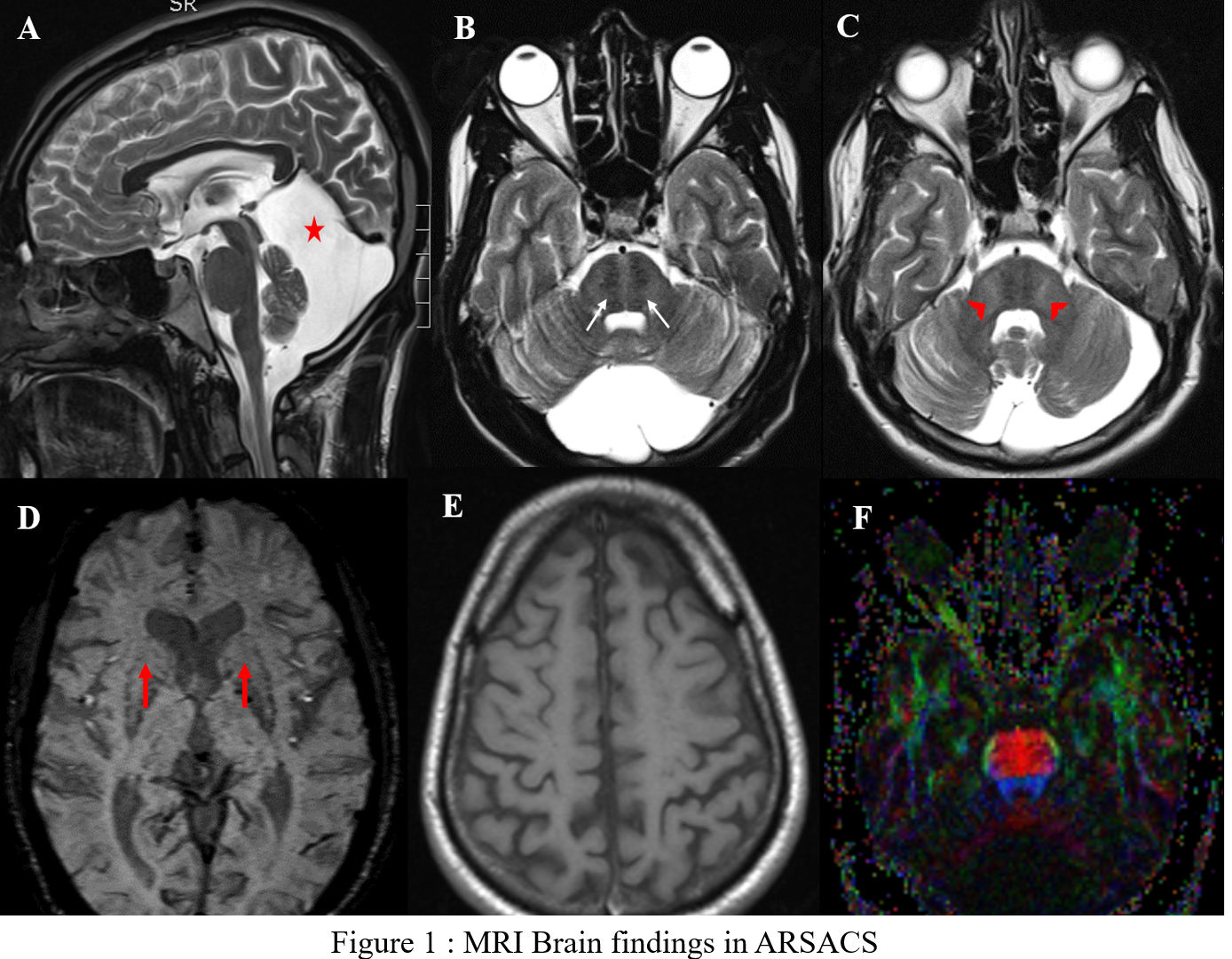
Results: 7 out of 9 patients were identified to have mutation in the SACS gene. Age at presentation ranged from 20 to 55 years (mean=29.8±11.9 years) with a mean disease duration of 12.7 years (SD-7.65, range 5-22 years). All patients (except 1) had symptom onset in the form of gait ataxia noticed < 20 years. Additional features were slow, hypometric saccades (1/7), seizures (1/7), kyphoscoliosis (1/7), dysmorphic facies (1/7) and subnormal intelligence (4/7). SDFS showed a score of 3 in 5/7 patients signifying moderate disability with independent ambulation. MRI (fig1) showed superior vermian & cerebellar atrophy (7/7),linear T2 hypointensities in pons(7/7),FLAIR hyperintensities where lateral pons merges with middle cerebellar peduncle(MCP) (7/7), thickening of MCP (3/7), bilateral lateral thalamic hyperintensities (6/7), posterior fossa arachnoid cyst (4/7) ,thinning of posterior mid body of corpus callosum (7/7), marginal mineralisation of the basal ganglia (7/7), bilateral parietal atrophy (7/7) and thinning of corticospinal tract on diffusion tensor imaging (DTI) (7/7). We identified 3 homozygous novel deletions chr13:23905161-23905161delCTCT, chr 13:001278055: exon8:c.8352delA and chr13:23909222delT resulting in frameshift mutations in 4 patients (including 2 siblings). 1 patient each had a homozygous nonsense mutation (chr13:23904883G>A- premature protein truncation) and homozygous deletion (chr13:23915575-23915576delAT) respectively which were previously identified as pathogenic.
Conclusion: This is the largest genetically confirmed series of ARSACS patients from India highlighting 3 homozygous novel mutations. MRI –DTI is a sensitive tool to identify pyramidal tract involvement which was present in all.
References: [1] Filho F M R, Parkinson. M.H,Pedroso J.L, Poh R, Faber I, Lourenco C.M, et al, Clinical, ophthalmological, imaging and genetic features in Brazilian patients with ARSACS. Parkinsonism and Related Disorders 62 (2019): 148–155.
[2] A. Duquette, B. Brais, J.P. Bouchard, J. Mathieu, Clinical presentation and early evolution of spastic ataxia of Charlevoix-Saguenay, Mov. Disord. 28 (2013) 2011–2014)
To cite this abstract in AMA style:
A. Cherian, K P. Divya, B. Thomas. Clinical, imaging and genetic characteristics from an Indian ARSACS cohort [abstract]. Mov Disord. 2022; 37 (suppl 2). https://www.mdsabstracts.org/abstract/clinical-imaging-and-genetic-characteristics-from-an-indian-arsacs-cohort/. Accessed March 27, 2026.« Back to 2022 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/clinical-imaging-and-genetic-characteristics-from-an-indian-arsacs-cohort/