Session Information
Date: Saturday, October 6, 2018
Session Title: Myoclonus
Session Time: 1:45pm-3:15pm
Location: Hall 3FG
Objective: To characterize clinically and genetically a series of 23 patients with myoclonus dystonia syndrome (MDS) and to explore the Unified Myoclonus Rating Scale (UMRS) to assess disease severity and evolution after bilateral deep brain stimulation of the internal globus pallidum (GPi-DBS).
Background: MDS is a genetic movement disorder characterized by dystonia and myoclonic jerks, often associated with psychiatric comorbidities. Common onset is on the first two decades of life. Daily life activities and quality of life might be affected, mainly due to gait impairment , feeding/drinking and writing difficulties. Pharmacological treatment has limited efficacy and poor tolerability. GPi-DBS has been described as an effective option with potentially long-term benefit.
Methods: Cross-sectional study of patients with MDS seen at a reference center for movement disorders in Spain. The UMRS was used to assess myoclonus severity. Genetic testing of SGCE mutations was performed by Sanger method.
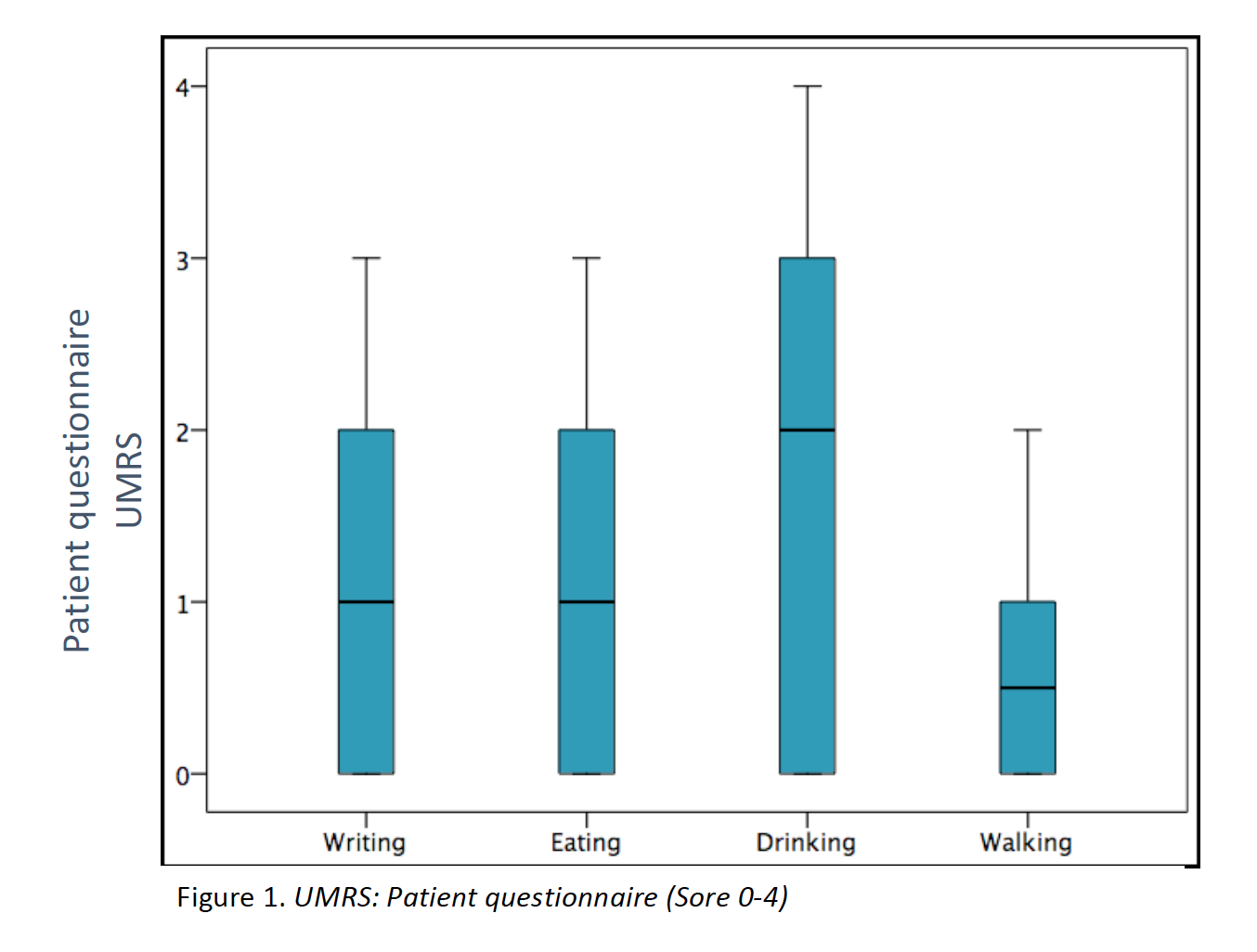
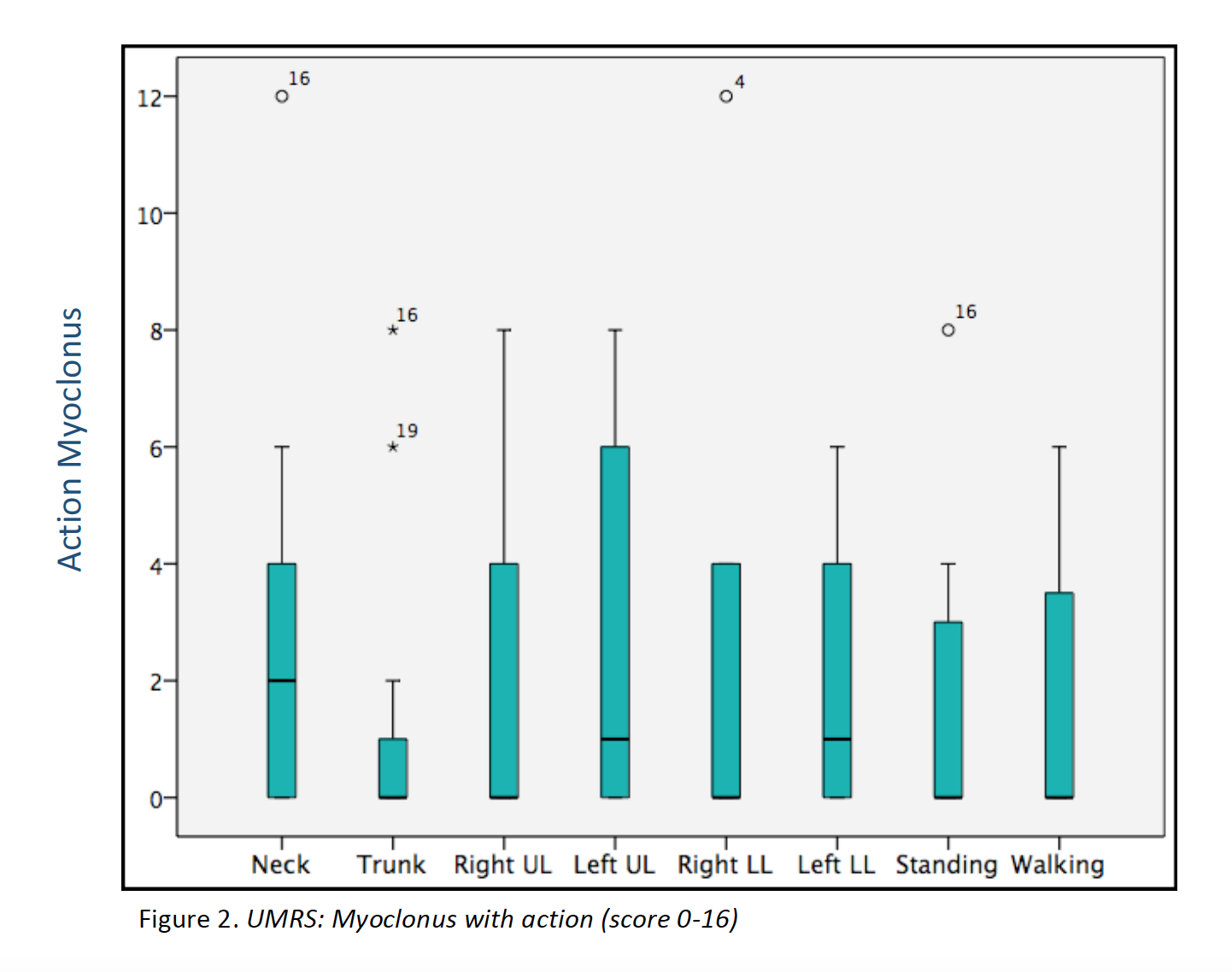
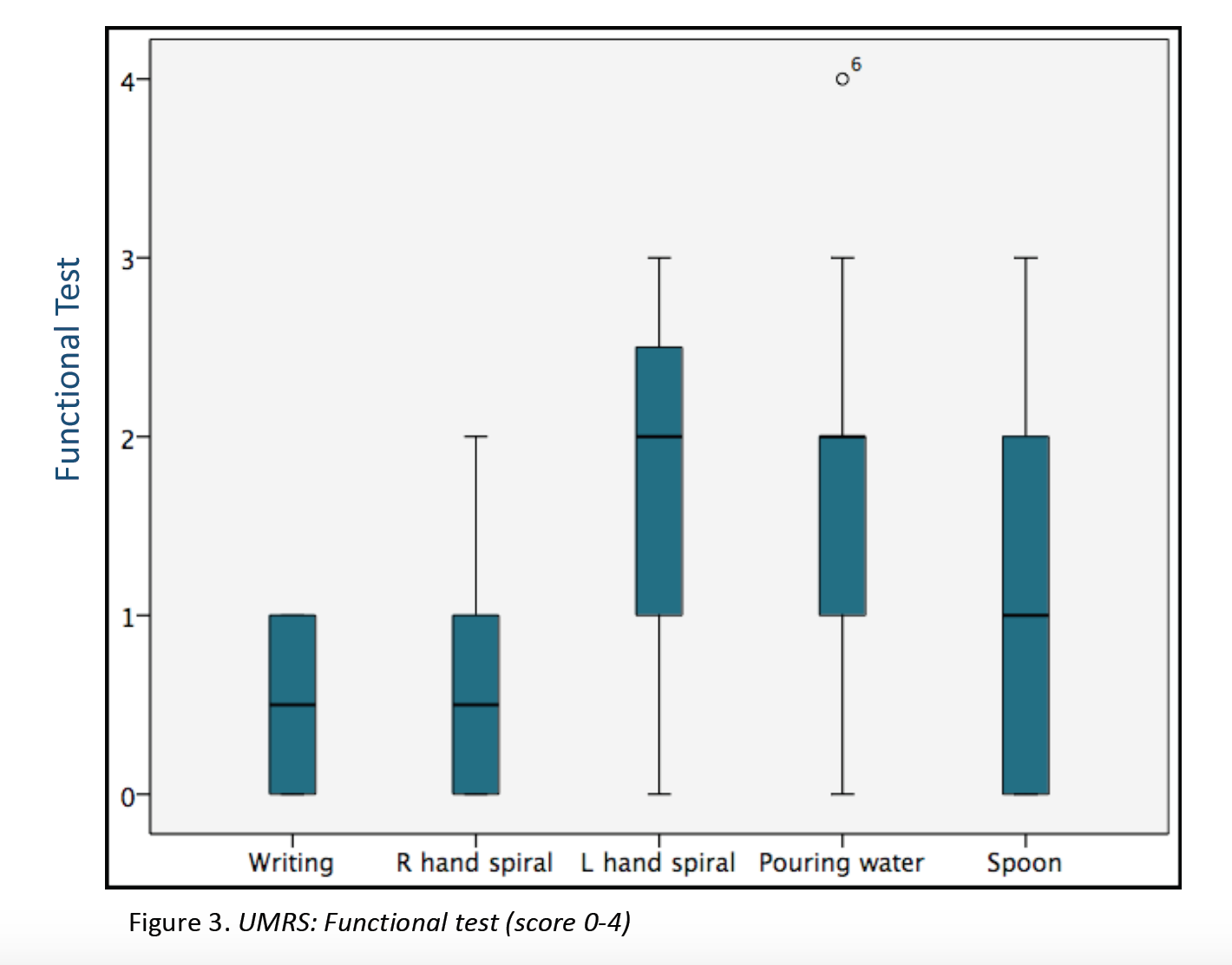
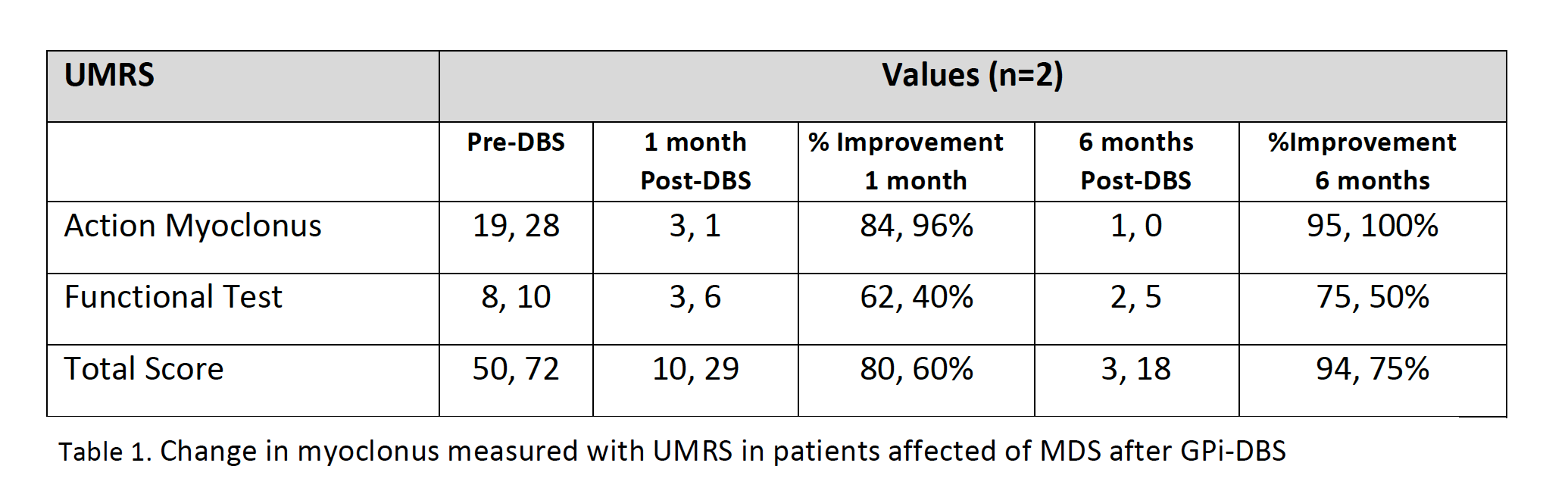
Results: 23 patients (11 males/12 females) from 16 families were included (median age at movement disorder onset: 2.7y, range 1-8y). Symptoms at debut were mainly myoclonus (n=16), followed by combination of myoclonus and dystonia (n=6). Mean age at clinical assessment was 12,7y (range 3-46y). Neuropsychiatric symptoms were observed in 14 patients, being anxiety the most frequent (n=12). UMRS was applied to 19 children with a mean age of 8,5y (range 3 -16y) and 4 adults with a median age of 29,2y (range 19-46y). Predominantly difficulties secondary to movement disorder, were reported in eating, drinking and walking (figure 1). Action myoclonus was observed predominantly in upper limbs, legs and neck (figure 2). In functional tests, patients showed more difficulties in Archimedes spiral drawing with left hand and pouring water (figure 3). SGCE mutations were identified in all patients. Genetic testing in 11 families identified the same mutation in 7 asymptomatic fathers. Two teenagers treated with GPi-DBS showed a significant reduction in action myoclonus and functional performance according to the UMRS (table 1).
Conclusions: Our series expands the clinical and genetic spectrum of MDS due to SGCE mutations. The UMDRS seems a valid method to assess myoclonus severity in children. The beneficial use of DBS in MDS is encouraging and should be indicated and further explored in the paediatric population.
References: 1. Myoclonus–Dystonia Syndrome: Clinical Presentation, Disease Course, and Genetic Features in 11 Families. Nardo Nardocci and cols. Movement Disorders Vol. 23, No. 1, 2008, pp. 28–34. 2. Inherited myoclonus dystonia . Asmus F, Gasser T.. Adv Neurol 2004; 94:113–119. 3. Primary Myoclonus-Dystonia: A Diagnosis Often Missed in Children Debabrata Ghosh and cols. Journal of Child Neurol. 2013 Nov; 28(11):1418-1422. 4. Jerky” dystonia in children: spectrum of phenotypes and genetic testing. Asmus and cols. Mov Disord. 2009 Apr 15;24(5):702-9. 5. Defining the epsilon-sarcoglycan (SGCE) gene phenotypic signature in myoclonus-dystonia: a reappraisal of genetic testing criteria. Carecchio M1, Magliozzi M, Copetti M, Ferraris A, Bernardini L, Bonetti M, Defazio G, Edwards MJ, Torrente I, Pellegrini F, Comi C, Bhatia KP, Valente EM. Mov Disord. 2013 Jun;28(6):787-94. 6.Psychiatric disorders, myoclonus dystonia and SGCE: an international study. Peall ans cols. Ann Clin Transl Neurol. 2015 Nov 20;3(1):4-11. 7. A missense mutation in KCTD17 causes autosomal dominant myoclonus-dystonia. Mencacci NE, Rubio-Agusti I, Zdebik A, Asmus F, Ludtmann MH, Ryten M et al. Am J Hum Genet. 2015 Jun 4;96(6):938-47. 8. Alteration of striatal dopaminergic neurotransmission in a mouse model of DYT11 myoclonus-dystonia. Zhang L1, Yokoi F, Parsons DS, Standaert DG, Li Y. PLoS One. 2012;7(3):e33669. 9. Abnormal nuclear envelopes in the striatum and motor deficits in DYT11 myoclonus-dystonia mouse models. Yokoi F1, Dang MT, Zhou T, Li Y. Hum Mol Genet. 2012 Feb 15;21(4):916-25. 10. Absolute quantification by droplet digital PCR versus analog real-time PCR. Hindson CM, Chevillet JR, Briggs HA, Gallichotte EN, Ruf IK, Hindson BJ, et al. Nature Methods 2013;10:1003–5. 11. The unified myoclonus rating scale. Frucht SJ, Leurgans SE, Hallett M, Fahn S. Adv Neurol 2002; 89:361–376. 12. Burke-Fahn-Marsden dystonia severity, Gross Motor, Manual Ability, and Communication Function Classification scales in childhood hyperkinetic movement disorders including cerebral palsy: a ‘Rosetta Stone’ study. Elze MC, Gimeno H, Tustin K, Baker L, Lumsden DE, Hutton JL, Lin JP. Dev Med Child Neurol. 2016 Feb;58(2):145-53. 13. Role of major and brain-specific Sgce isoforms in the pathogenesis of myoclonus-dystonia syndrome. Xiao J, Vemula SR, Xue Y, Khan MM, Carlisle FA, Waite AJ, Blake DJ, Dragatsis I, Zhao Y, LeDoux MS. Neurobiol Dis. 2017 Feb; 98:52-65. 14. Faithful SGCE imprinting in iPSC-derived cortical neurons: an endogenous cellular model of myoclonus-dystonia. Grütz K, Seibler P, Weissbach A, Lohmann K, Carlisle FA, Blake DJ, Westenberger A, Klein C, Grünewald A. Sci Rep. 2017 Feb 3;7:41156. 15. Deep Brain Stimulation in Rare Inherited Dystonias. Beaulieu-Boire I, Aquino CC, Fasano A, Poon YY, Fallis M, Lang AE et al. Brain Stimul 9(6):905-910, 2016. 16. Deep Brain Stimulation in Myoclonus-Dystonia Syndrome. Cif L, Valente EM, Hemm S, Coubes C, Vayssiere N, Serrat S, et al. Mov Disord 19(6):724-7, 2004. 17. Factors predicting protracted improvement after pallidal DBS for primary dystonia: the role of age and disease duration. Isaias IU, Volkmann J, Kupsch A, Burgunder JM, Ostrem JL, Alterman RL, et al: J Neurol 258:1469-1476, 2011. 18. Deep brain stimulation for childhood dystonia: is ‘where’ as important as in ‘whom’. Lumsden DE, Kaminska M, Ashkan K, Selway R, Lin JP: . Eur J Paediatr Neurol 21(1):176-184, 2017. 19. Cognitive function in children with primary dystonia before and after deep brain stimulation. Owen T, Gimeno H, Selway R, Lin JP. Eur J Paediatr Neurol 19(1):48-55, 2015. 20. Surgical Treatment of Myoclonus Dystonia Syndrome. Rughani AI, Lozano AM. Mov Disord 28(3):282-287, 2013.
To cite this abstract in AMA style:
B. Perez-Dueñas, M. Vanegas, L. Marti, A. Darling, D. Ortigoza-Escobar, S. Candela, H. Baide, J. Campistol, S. Aguilera, M. Marti, A. Macaya. Clinical and genetics characterization of patients with Myoclonus Dystonia Syndrome [abstract]. Mov Disord. 2018; 33 (suppl 2). https://www.mdsabstracts.org/abstract/clinical-and-genetics-characterization-of-patients-with-myoclonus-dystonia-syndrome/. Accessed March 29, 2026.« Back to 2018 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/clinical-and-genetics-characterization-of-patients-with-myoclonus-dystonia-syndrome/