Session Information
Date: Monday, September 23, 2019
Session Title: Rare Genetic and Metabolic Diseases
Session Time: 1:45pm-3:15pm
Location: Les Muses Terrace, Level 3
Objective: We aim to investigate the clinical and genetic features of PKD in a large cohort of patients in with WES.
Background: PKD is a rare movement disorder characterized by brief attacks of dystonia and/or choreoathetosis triggered by sudden movements. The first causative gene, PRRT2 was identified in 2011 [1]. However, approximately 20% of familial cases and 65% of sporadic cases of PKD remain genetically unexplained [2]. Recent advances in next generation sequencing has resulted in an explosion in the genes associated with PKD, including SCN8A,DEPDC5,SCL2A1.
Method: Patients were recruited from Singapore between 2002 and 2018 and Malaysia in 2016. Subjects had been diagnosed with PKD according to established clinical criteria underwent a standardized interview to characterize the movement disorder Venous blood samples were collected, and DNA extracted for WES was performed. Genes known to be associated with the paroxysmal movement disorders were screened. Analysis of sequences was performed to determine potential pathogenic mutations.
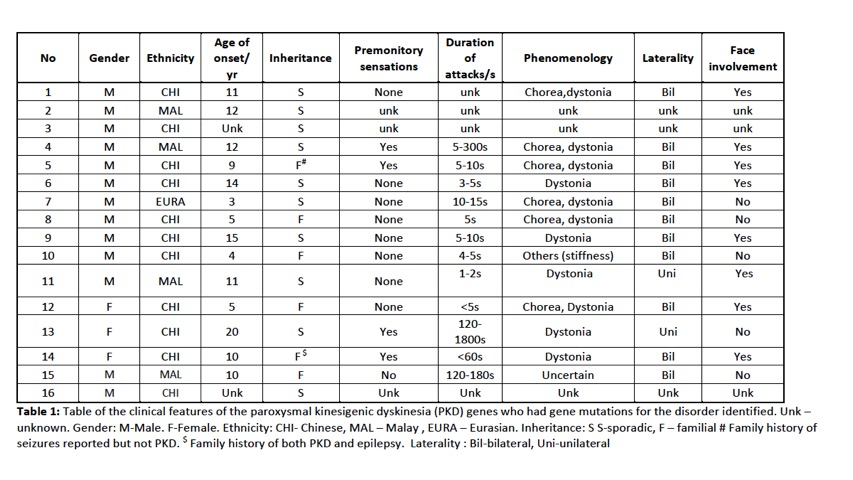
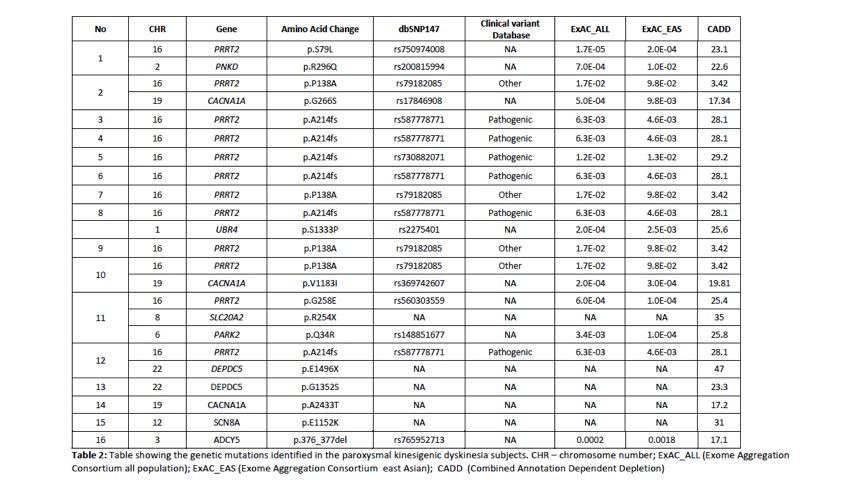
Results: A total of 88 subjects were included in the study, of which there were 54 index cases where genetic testing was performed. 29 probands had earlier been screened using targeted gene sequencing: 15 were identified to be PRRT2 positive and 14 PRRT2 negative. A total of 39 probands underwent WES. Of these 39 probands, 12 were identified to have PRRT2mutations including 2 subjects with previously unreported novel point mutations in PRRT2. Four subjects had mutations identified in other rarer gene mutations known to be associated with PKD : 2 subjects had mutations in DEPDC5and 1 subject each with SLC20A2and SCN8A. Multiple gene mutations were identified in 6 subjects (see Table 2). Clinical information is presented in Table 1. In the remaining 23 subjects,mutations in the genes known to be associated with the paroxysmal movement disorders were not identified.
Conclusion: In PKD, PRRT2remains the most common genetic mutation identified. We confirm in our study that DEPDC5, SLC20A2 and SCN8Amutations can also be found in our population and are probably not as uncommon causes of PKD. We also report patients with multiple gene mutations. The use of WES has enabled the detection of mutations associated with PKD, and developments in NGS will enable better genetic phenotyping of this disorder.
References: 1. Chen, W.J., et al., Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet, 2011. 43(12): p. 1252-5. 2. McGovern, E.M., E. Roze, and T.J. Counihan, The expanding spectrum of paroxysmal movement disorders: update from clinical features to therapeutics. Curr Opin Neurol, 2018. 31(4): p. 491-497.
To cite this abstract in AMA style:
Z. Xu, Z. Lu, CK. Lim, SC. Low, E. Ng, AH. Tan, SY. Lim, EK. Tan, LCS. Tan. Clinical and genetic features of paroxysmal kinesigenic dyskinesia (PKD) studied with whole exome sequencing (WES) [abstract]. Mov Disord. 2019; 34 (suppl 2). https://www.mdsabstracts.org/abstract/clinical-and-genetic-features-of-paroxysmal-kinesigenic-dyskinesia-pkd-studied-with-whole-exome-sequencing-wes/. Accessed March 17, 2026.« Back to 2019 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/clinical-and-genetic-features-of-paroxysmal-kinesigenic-dyskinesia-pkd-studied-with-whole-exome-sequencing-wes/