Session Information
Date: Monday, October 8, 2018
Session Title: Parkinson's Disease: Pathophysiology
Session Time: 1:15pm-2:45pm
Location: Hall 3FG
Objective: We report the first case of myotonic dystrophy (DM) type 1 with DMPK mutations presenting with parkinsonian syndrome.
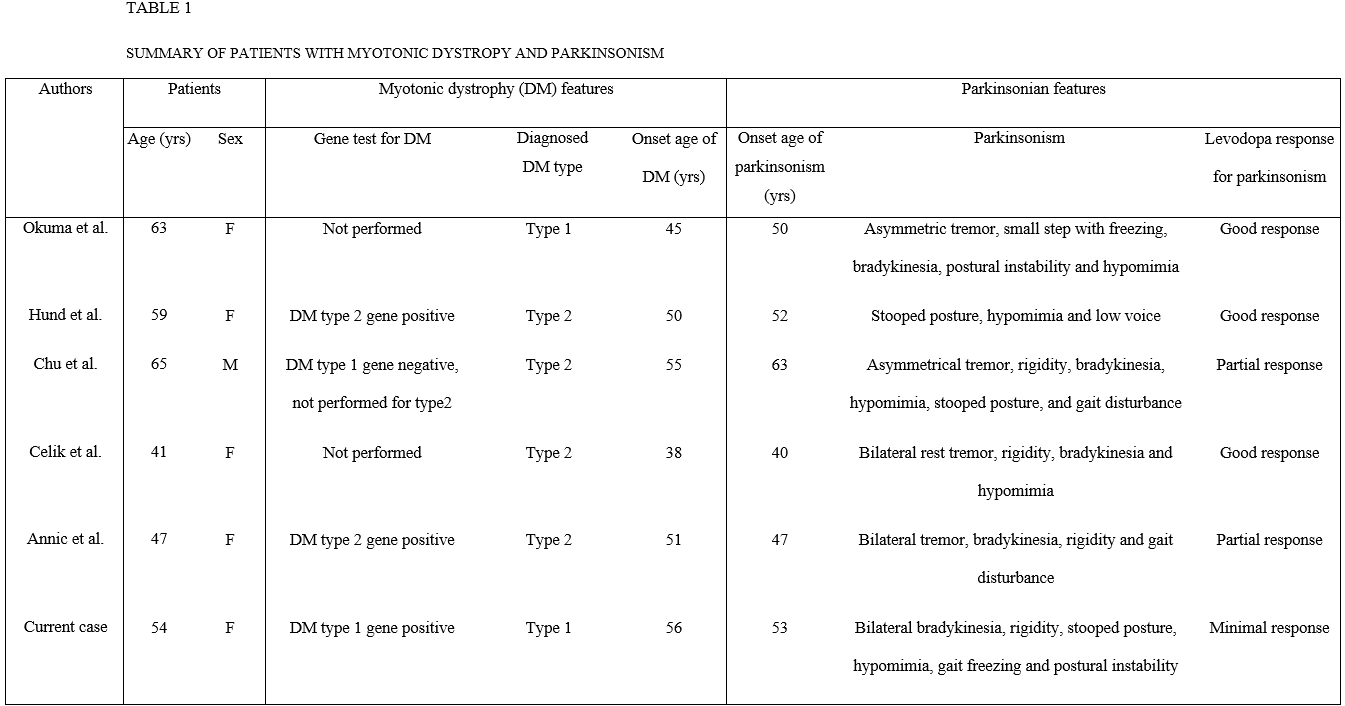
Background: DM type 1 is a genetic disorder with muscle dystrophy and myotonia in combination with multisystem involvement of the eye, heart, endocrine system, and central nervous system (CNS). There were five reports of DM with parkinsonism; four reports of DM type 2 and one report of clinically suspected genetically unproven DM type 1 [Table1].
Methods: Case report.
Results: A 54-year-old woman presented with one year history of muscle stiffness and uncomfortable range of motion in four limbs. On examination, she had a mild to moderate bilateral bradykinesia, mild axial and bilateral limb rigidity, mild stooped posture and hypomimia. She had no resting and kinetic tremor. Eye movements showed breaking up of smooth pursuit but no supranuclear gaze palsy. Autonomic symptoms and signs were not evident. She did not have depression, sleep problems and loss of smell. The mini mental state examination score was 29/30 and the clinical dementia rating score was 0/5. MRI showed mild diffuse cerebral atrophy. A levodopa trial showed subjective minimal improvement. The initial diagnosis was atypical parkinsonian syndrome based on bilateral symmetric parkinsonian features and poor levodopa response. FP-CIT PET showed moderate to severe decrease in bilateral putaminal uptake. At age 56, she developed neck weakness and swallowing difficulty. Examination showed mild bifacial weakness, and percussion and grip myotonia. Needle electromyography confirmed myotonic discharges in proximal and distal muscles. Ophthalmologic examination showed bilateral cataract. A gene study of the DM Type 1 showed an expanded CTG repeat length with 120 repeats in the DMPK gene. At age 58, there were gait freezing, postural instability and then frequent falls as well as moderate stooped posture. Increasing doses of levodopa with 1300mg per day were ineffective. She developed dysarthria, dysphasia and severe weight loss. At age 59, she died from asphyxia.
Conclusions: Several autopsy findings in patients with clinically diagnosed DM and CNS symptoms showed intracytoplasmic inclusion bodies (IIBs) in the substantia nigra (SN), thalamus, caudate nucleus, putamen and cortex. These IIBs were different from inclusions in the muscle which consisted with expanded repeat mutant RNA in patients with DM. IIBs may possibly represent pathogenesis of CNS symptoms. Higher load of IIBs in the SN and striatum may cause parkinsonism in patients with DM.
References: 1. Ono S, Inoue K, Mannen T, Mitake S, Shirai T, Kanda F, et al. Intracytoplasmic inclusion bodies of the thalamus and the substantia nigra, and Marinesco bodies in myotonic dystrophy: a quantitative morphological study. Acta neuropathologica. 1989;77(4):350-6. 2. Ono S, Takahashi K, Fukuoka Y, Jinnai K, Kanda F, Kurisaki H, et al. Intracytoplasmic inclusion bodies of the substantia nigra in myotonic dystrophy. Immunohistochemical observations. Journal of the neurological sciences. 1997;148(2):193-8.
To cite this abstract in AMA style:
J. Choi, J. Lee, H. Kim, B. Jeon. A patient with myotonic dystrophy type 1 and parkinsonism [abstract]. Mov Disord. 2018; 33 (suppl 2). https://www.mdsabstracts.org/abstract/a-patient-with-myotonic-dystrophy-type-1-and-parkinsonism/. Accessed April 20, 2025.« Back to 2018 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/a-patient-with-myotonic-dystrophy-type-1-and-parkinsonism/