Category: Rare Genetic and Metabolic Diseases
Objective: To study the spectrum of movement disorders phenomenologies (MDs) in genetically proven ceroid lipofuscinosis neuronal (CLNs)
Background: MDs are increasingly recognized in the CLNs with spectrum ranging from myoclonus, ataxia, dystonia to parkinsonism[1]
Method: A single-centre retrospective study of patients of genetically proven CLNs with MDs at the onset or during the course of the illness
Results: Eight patients (5 males) of genetically proven CLNs with MDs were included. The median (range) age at onset was 9.5 years (1-18) and duration of illness was 2 years (0.5-3). None had a positive family history while 4 had consanguineous parentage. The most common symptom at onset was walking difficulty (4 patients). History suggestive of cerebellar abnormality was noted in 5 patients, myoclonus in 2, dystonia in 2 and parkinsonism in 1. On examination, cognition was impaired in 4 patients, abnormal eye movements in 5 patients, dysarthria in 6 patients, and pyramidal signs in 3 patients. Cerebellar abnormality and dystonia (generalized truncal predominant dystonia in 3 and focal leg dystonia in 1) was the most frequent MDs seen in 4 patients (50%) each followed by multifocal distal predominant myoclonus in 3 patients and parkinsonism in 2 patients. MRI brain [Figure-1] was abnormal in 6 patients (75%, cerebellar atrophy in 4 patients, hypomyelination with T2 hypointense thalamus and cerebral atrophy with T2 hypointense thalamus in 1 each). Exome sequencing revealed biallelic pathogenic or likely pathogenic variants in all 8 patients: CLN1 (PPT1) in 1 patient (homozygous c.674T>C;p.Phe225Ser variant), CLN2 (TPP1) in 4 (homozygous c.1069G>A;p.Ala357Thr, c.622C>T;p.Arg208Ter, and c.1679T>C;p.Leu159Pro variants and compound heterozygous c.1190delT;p.Phe397SerfsTer30 and c.1435C>G;p.Pro479Ala variants in 1 each), CLN6 (CLN6) in 1 patient (compound heterozygous c.298-1G>C and c.476C>T;p.Pro159Ala) and CLN12 (ATP13A2) in 2 patients (homozygous c.705G>C;p.Glu235Asp and c.2218C>T;p.Arg740Ter in 1 patient each). 2/4 patients with CLN2 had spinocerebellar ataxia autosomal recessive type-7 presentation. Both patients with CLN12 (ATP13A2) had dystonia-parkinsonism presentation.
Conclusion: Ataxia is the most frequent movement disorder phenomenology noted in CLN. CLN2 was the most frequent subtype among CLNs with movement disorders.
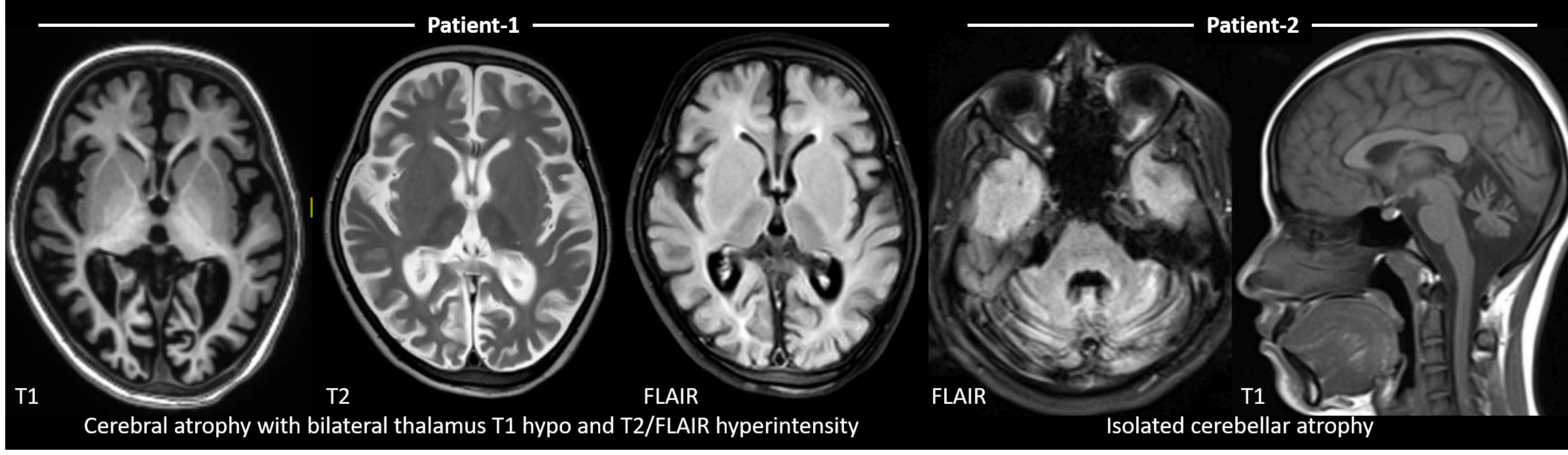
MRI brain of Patient-1 (CLN1) and Patient-2 (CLN2)
References: [1] A. Simonati, R.E. Williams, Neuronal ceroid lipofuscinosis: the multifaceted approach to the clinical issues, an overview, Frontiers in neurology 13 (2022) 811686.
To cite this abstract in AMA style:
VV. Holla, N. Kamble, G. Arunachal, B. Muthusamy, R. Yadav, PK. Pal. Clinical, Radiological and Genetic profile of Eight Patients with Genetically Proven Ceroid Lipofuscinosis Neuronal and Movement Disorders [abstract]. Mov Disord. 2024; 39 (suppl 1). https://www.mdsabstracts.org/abstract/clinical-radiological-and-genetic-profile-of-eight-patients-with-genetically-proven-ceroid-lipofuscinosis-neuronal-and-movement-disorders/. Accessed June 15, 2026.« Back to 2024 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/clinical-radiological-and-genetic-profile-of-eight-patients-with-genetically-proven-ceroid-lipofuscinosis-neuronal-and-movement-disorders/