Category: Parkinson's Disease: Genetics
Objective: Functional validation of rare variants of lysosomal genes potentially associated with early onset Parkinson’s disease (EOPD)
Background: The genetic load in EOPD is high; however, the yield of genetic diagnosis in these patients is still very low. Genomic approaches are allowing the identification of new candidate genes that could be associated with EOPD, either Mendelian genes or risk factors that could act as facilitators of disease expression. Given that dysfunction of the lysosomal pathway can facilitate the development of PD (1), in this work we propose that other genes involved in this pathway could form part of the genetic architecture of EOPD. Here we show experimental evidence linking lysosomal genes and EOPD expression
Method: We used precision phenotyping and rating scales for the diagnosis of PD (UK Brain Bank Criteria). We performed trio-WES or singleton WES for genomic analysis in 41 EOPD clinical series. A custom bioinformatic pipeline was used for variants filtering in a list of genes. Functional studies were in patients’ fibroblasts using computational super-resolution microscopy (CSRM) analysis
Results: We found two EOPDs males carrying p.Asp313Tyr in the X-linked GLA gene (encodes α-galactosidase) of Fabry disease. Another patient (male) was a heterozygous carrier of p.Arg419Gln in GLB1 (encodes β-galactosidase) related to the autosomal recessive diseases GM1 gangliosidosis types I-IV and mucopolysaccharidosis type IVB. None of these findings allowed the genetic diagnosis of the patients; however, experimental studies did yield pieces of evidence of lysosomal dysfunction.
α-galactosidase and β-galactosidase are found in both lysosomes and the Golgi apparatus (GA). We found in fibroblasts carrying GLA:p.Asp313Tyr a deficiency of α-galactosidase and partial retention in the GA. Fibroblasts carrying GLB1:p.Arg419Gln showed no β- galactosidase expression differences and GA abnormalities.
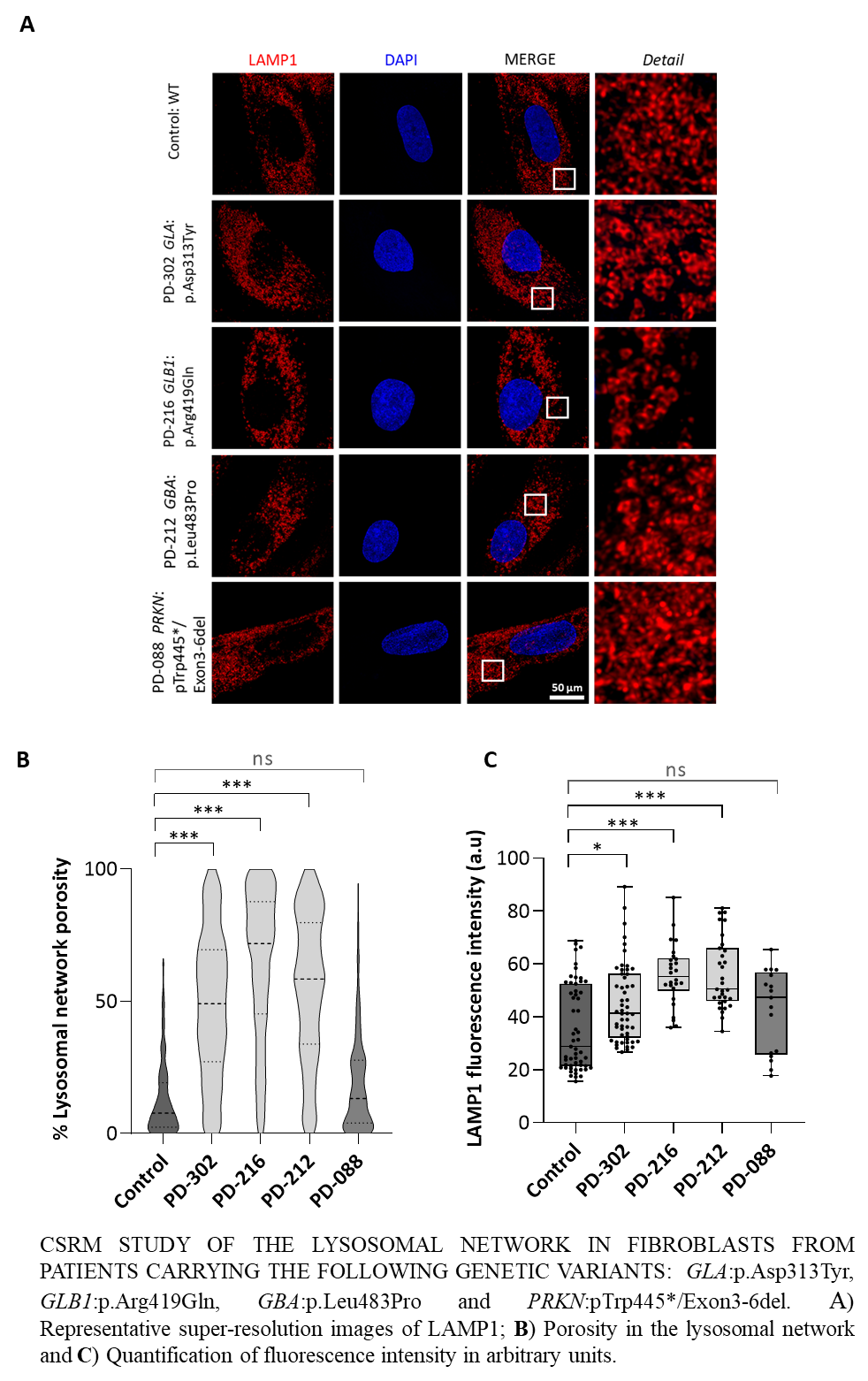
Notably, comparison by CSRM of GLA:p.Asp313Tyr, GLB1:p.Arg419Gln, GBA:p.Leu483Pro and PRKN:pTrp445*/Exon3-6del fibroblasts showed higher expression levels of the lysosomal marker LAMP1 and lysosomal network defects in all but PRKN:pTrp445*/Exon3-6del (Figure)
Conclusion: These results highlight the relevance of lysosomal dysfunction in the etiology of EOPD and the need for functional genomics to understand the pathophysiology of this group of disorders
Figure
References: L.A. Robak, I.E. Jansen, J. vanRooij, A.G. Uitterlinden, R. Kraaij, J. Jankovic, P. Heutink, J.M. Shulman.
International Parkinson’s Disease Genomics Consortium (IPDGC), Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease
Brain, 140 (2017), pp. 3191-3203
To cite this abstract in AMA style:
J. Hoenicka, A. Pascual, O. de Fàbregues, M. Frias, L. Vela, M. de Lucca, P. García-Ruiz, C. Feliz, M. Marchen, R. Repossi, G. Fernández, M. Roldán, F. Palau. Lysosomal network defects in parkinsonian patients carrying rare variants in lysosomal hydrolytic enzyme genes [abstract]. Mov Disord. 2024; 39 (suppl 1). https://www.mdsabstracts.org/abstract/lysosomal-network-defects-in-parkinsonian-patients-carrying-rare-variants-in-lysosomal-hydrolytic-enzyme-genes/. Accessed March 31, 2025.« Back to 2024 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/lysosomal-network-defects-in-parkinsonian-patients-carrying-rare-variants-in-lysosomal-hydrolytic-enzyme-genes/