Category: Rare Genetic and Metabolic Diseases
Objective: To describe 2 monozygotic twins carrying biallelic ECHS1 mutations with different clinical phenotypes
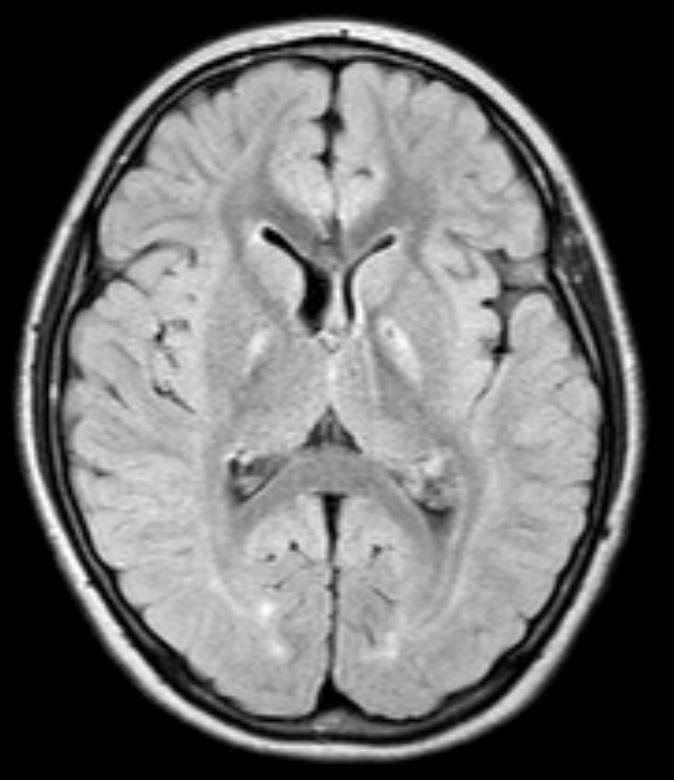
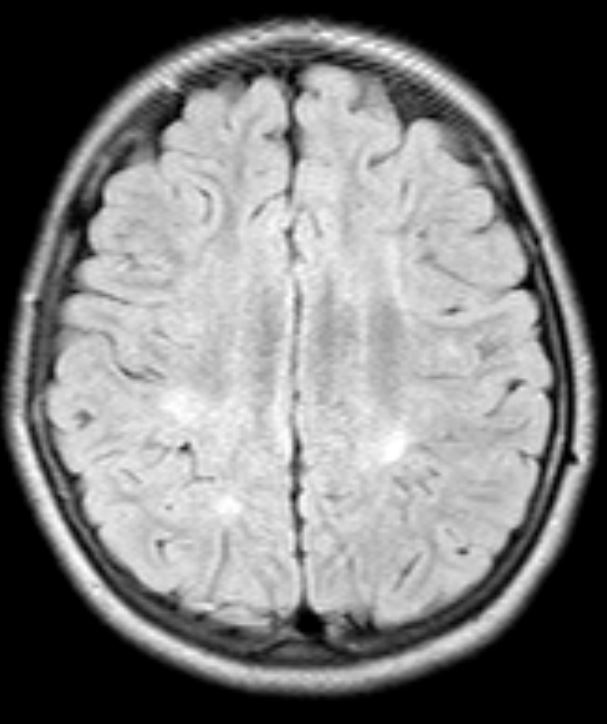
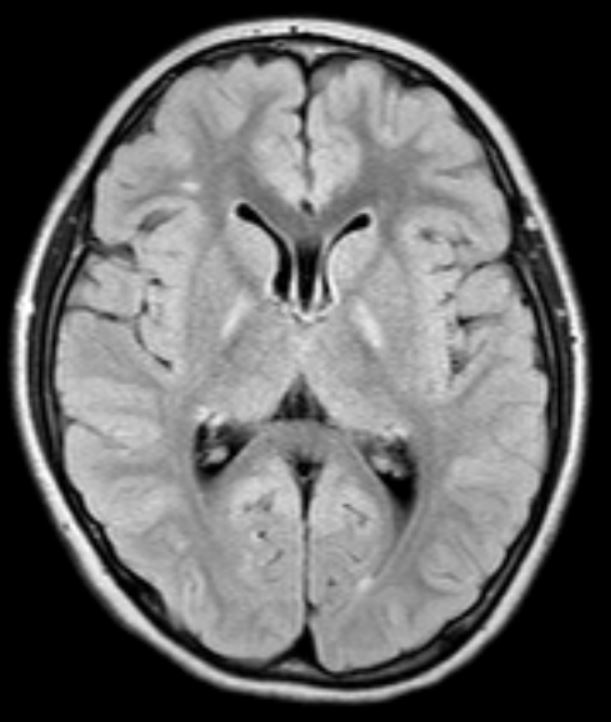
Background: ECHS1 encodes for a mitochondrial short chain enoyl-CoA hydratase, a key component of degradation of fatty acids and essential amino acids. Biallelic mutations are associated with a rare congenital neurometabolic disorder with a heterogeneous clinical spectrum, featuring severe developmental delay, psychomotor regression, dystonia and seizures; paroxysmal dyskinesia has rarely been reported. Onset is classically in childhood, presentation after 8 years has not been documented so far. Brain MRI usually shows subcortical white matter alterations and pallidal involvement with Leigh-like features
Method: We describe 2 monozygotic twin girls aged 16 with clinical assessment, MRI imaging and genetic testing
Results: The first twin presented mild developmental delay and paroxysmal episodes of generalized painful dystonia from 18 months of age. Dystonic attacks started in the lower limbs with a caudo-cranial spreading to the trunk, neck and upper limbs leading to opisthotonic posturing and head hyperextension. Triggers included prolonged physical exercise, temperature changes and hot water; duration was up to one hour and remission was favored by rest. Carbamazepine significantly reduced frequency, severity and anatomical extension of the attacks. Mild intellectual disability (IQ53) was documented during adolescence. At age 14 years, the second twin started complaining of mild episodes of exercise-induced foot dystonia, with occasional painful involvement of the homolateral upper limb. Cognition was normal. Carbamazepine abolished the attacks. Genetic analysis (WES) demonstrated heterozygous mutations in ECHS1 gene: c.518C>T p.Ala173Val (previously reported as pathogenic in 4 subjects with early-onset paroxysmal dyskinesia) and c.123_124delAG p.Gly42Glufs*3 (predicted as pathogenic) in both twins. Parents carried a single mutation. Brain MRI showed bilateral pallidal hyperintensity and white matter subtle alterations in both twins.
Conclusion: ECHS1 mutations can rarely cause a mild phenotype of paroxysmal dystonia with a later onset than previously reported and partial overlap with Glut-1 deficiency. The p.Ala173Val mutation is specifically associated with this clinical presentation. Our cases demonstrate intrafamilial clinical heterogeneity even in monozygotic twins with the same radiological alterations on brain MRI
To cite this abstract in AMA style:
G. Bonato, M. Nosadini, S. Andretta, A. Suppiej, A. Leon, S. Sartori, M. Carecchio. Paroxysmal dystonia with phenotypic variability in ECHS1 mutation twin carriers [abstract]. Mov Disord. 2022; 37 (suppl 2). https://www.mdsabstracts.org/abstract/paroxysmal-dystonia-with-phenotypic-variability-in-echs1-mutation-twin-carriers/. Accessed April 1, 2025.« Back to 2022 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/paroxysmal-dystonia-with-phenotypic-variability-in-echs1-mutation-twin-carriers/