Session Information
Date: Monday, October 8, 2018
Session Title: Parkinson's Disease: Genetics
Session Time: 1:15pm-2:45pm
Location: Hall 3FG
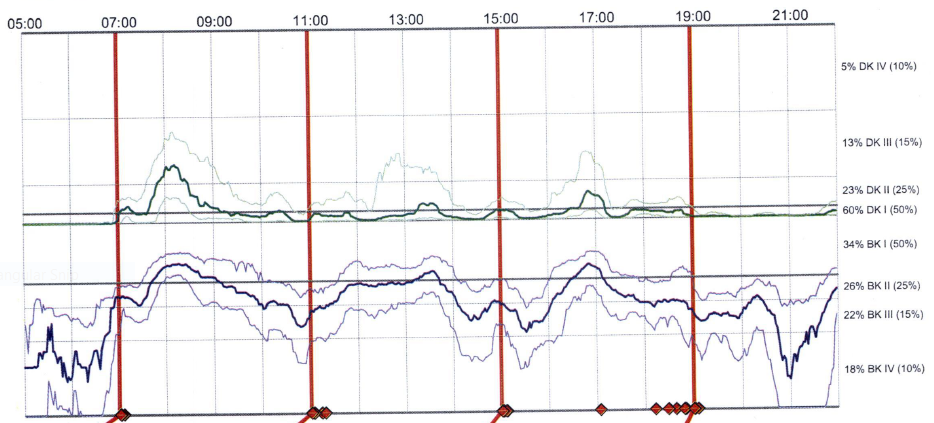
Objective: A 38 year old Australian florist of Anglo-Celtic origin described two years of difficulty using his right hand with associated sensory disturbance attributed initially to ulnar nerve pathology. After two failed ulnar nerve transposition surgeries and evolution of his problem with additional features of tremor, rigidity and bradykinesia, his diagnosis was revised to Idiopathic Parkinson’s disease. Examination by an experienced movement disorder neurologist documented right sided hemiparkinsonism with extrapyramidal rigidity and bradykinesia and no cerebellar features. An MRI of the brain was normal. He was commenced on levodopa therapy and experienced a dramatic improvement. Within six months of starting low dose levodopa therapy (100/25mg three times daily of Levodopa/Carbidopa) he demonstrated motor fluctuations, supported by Parkinson’s Kinetograph (figure 1). (Fig1) In addition to his motor symptoms he was managed for insomnia with melatonin and described mood fluctuations. There were no dysautonomic features and anosmia was not demonstrated. His 7 year old daughter at the age of 4 years was investigated for an ataxic syndrome including opsoclonus myoclonus that evolved following a viral illness. She was found to have a deletion in Chromosome region 3p26.2p26.1, the deleted region included the IPTR1 gene. Haploinsufficiency of IPTR1 has previously been reported in SCA15 (Leemput.J et al 2007). Our patient was subsequently shown to have the same deletion.
Background: SCA 15 was originally described in 2001 in an Australian family of Anglo-Celtic origin (Storey.E et al 2001). This family all demonstrated autosomal dominant inheritance and displayed cerebellar features. These patients typically presented with gait ataxia and progressed slowly and variably with truncal and limb ataxia, dysarthria, dysphagia, and ocular dysmetria . Postural tremor was noted to occur infrequently. Onset of symptoms varied widely between ages seven and 70 years and progression occurred over decades. MRI brain showed atrophy of the cerebellar vermis in some of these individuals. Synofzik et al. reported five SCA15 families, and four had deletions involving both genes, but one family’s deletion included only ITPR1 (Synifzik et al 2011). Since that time, all reported SCA15 families have had deletions involving ITPR1 thus establishing it as the causative gene of SCA15 (Tipson.P et al). Complete deletion of ITPR1 causes loss of mRNA transcription and reduced ITPR1 protein. ITPR1 protein’s primary function is to mediate the release of intracellular stores of calcium from the endoplasmic reticulum by playing a key role in the inositol messenger pathway. ITPR1 is highly expressed in the cerebellum Purkinje cells and other neurons. The slow degeneration of these cells may, therefore, be due to particular sensitivity to calcium homeostasis (Leemput et al 2007) SCA3 (Machado-Joseph disease) was the first SCA in which levodopa responsiveness was described (Park.H et al 2014). This has since been described relatively frequently in SCA2 and SCA17 (Park.H et al 2014). The clinical features of Parkinsonism in these disorders can be similar to idiopathic Parkinson’s disease, although they often have additional features mimicking multisystem atrophy. Patients with SCA17 can also have additional chorea, epilepsy and cognitive impairment. These patients have exhibited levodopa responsiveness and motor fluctuations with levodopa therapy.
Methods: N/A
Results: N/A

Conclusions: Our case has a novel phenotype for SCA15 and to our knowledge is the first case to be reported with a pure parkinsonian phenotype. Levodopa responsiveness in this patient is consistent with parkinsonism reported in other spinocerebellar ataxias.
References: 1.Storey E, A new autosomal dominant pure cerebellar ataxia. Neurology: Wolters Kluwer Health, Inc; 2001. 2.Leemput JVD, at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLOS GENETICS2007. 3.Park H, Parkinsonism in Spoinocerebellar Ataxia. BioMed Research International2014. 4. Tipton PW, Spinocerebellar ataxia 15: A phenotypic review and expansion. Neurologia i Neurochirurgia Polska; 2017. p. 86-91.
To cite this abstract in AMA style:
M. Ghaly, C. Wools, A. Evans, E. Storey. Daughter’s ataxia reveals father’s genetic Parkinsonism. The first reported case of Spinocerebellar ataxia -15 presenting with levodopa responsive hemi Parkinsonism [abstract]. Mov Disord. 2018; 33 (suppl 2). https://www.mdsabstracts.org/abstract/daughters-ataxia-reveals-fathers-genetic-parkinsonism-the-first-reported-case-of-spinocerebellar-ataxia-15-presenting-with-levodopa-responsive-hemi-parkinsonism/. Accessed April 1, 2025.« Back to 2018 International Congress
MDS Abstracts - https://www.mdsabstracts.org/abstract/daughters-ataxia-reveals-fathers-genetic-parkinsonism-the-first-reported-case-of-spinocerebellar-ataxia-15-presenting-with-levodopa-responsive-hemi-parkinsonism/